One destroys the factory. The other ignores its output. Know the antibodies, the genetics, and the organs that never needed insulin in the first place.
A 22-year-old woman presents to the ER with fruity-smelling breath, altered mental status, diffuse abdominal pain, and blood glucose of 480 mg/dL. She reports 3 weeks of polyuria, polydipsia, and a 12-pound weight loss. ABG shows pH 7.18, and urine shows ketones 3+. She has no family history of diabetes.
Which antibody is most commonly tested to confirm the underlying pathophysiology?
THE PATTERN
Two Diseases, One Name
Type 1 destroys the factory. Type 2 ignores its product.
Type 1: Destruction
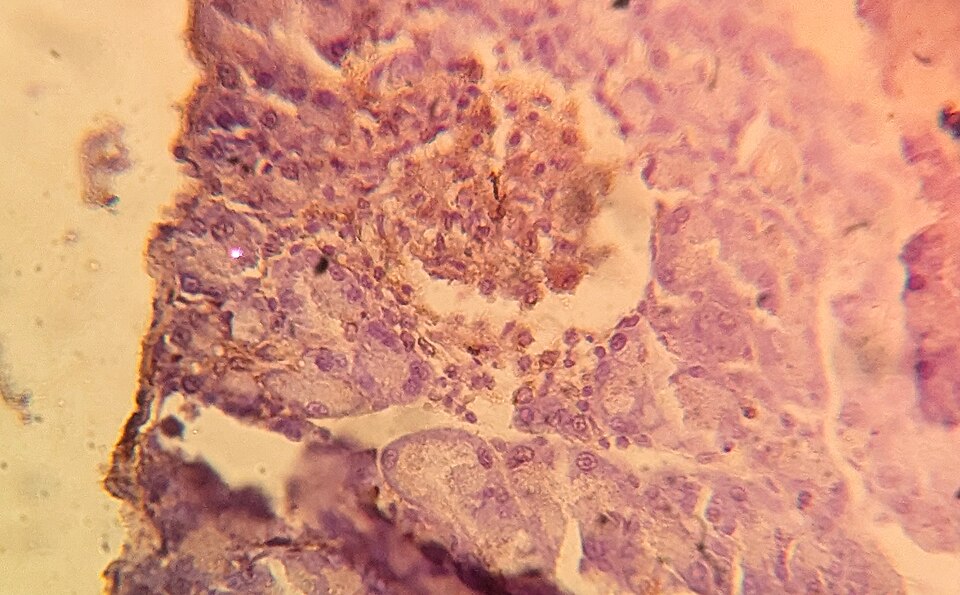
A pancreatic islet stained for insulin, showing the beta cells that autoantibodies destroy in Type 1 diabetes. Tap to expand.
HLA: DR3 and DR4💡DR3 and DR4: "age 3 and 4" is when Type 1 usually peaks in kids. The HLA matches the age.
Absolute insulin deficiency: zero production
Presents with DKA (fruity breath, Kussmaul breathing, abdominal pain)
Thin patient, weight loss, young onset
Second messenger: tyrosine kinaseInsulin receptor is a receptor tyrosine kinase. When insulin binds, it autophosphorylates and triggers GLUT4 translocation. This is the receptor that is WORKING in Type 1, but there is no insulin to activate it. (the receptor works, the hormone is missing)
Type 2: Resistance
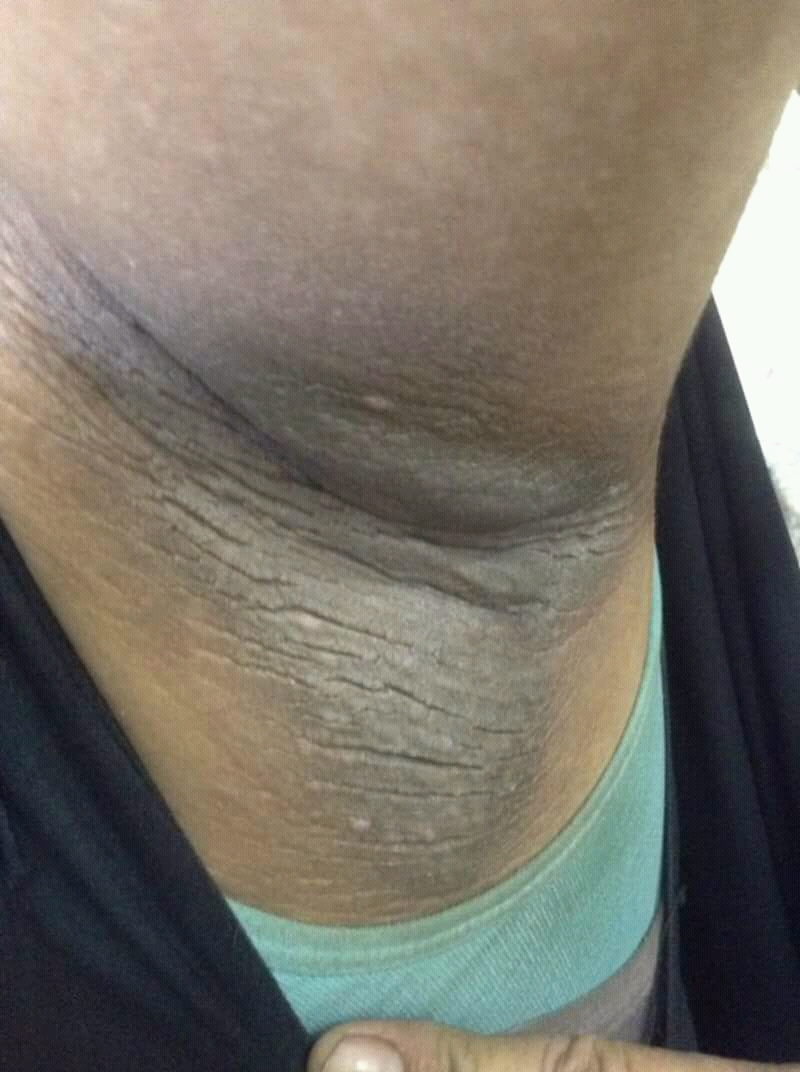
Velvety darkening of the neck folds that signals insulin resistance and points toward Type 2 diabetes. Tap to expand.
Insulin resistance in peripheral tissues
No autoimmune antibodies
Strong genetic component (concordance >90% in twins)
Relative insulin deficiency: beta cells produce insulin, tissues ignore it
Enough insulin to prevent ketosisEven a small amount of insulin suppresses lipolysis and prevents the liver from making ketone bodies. Type 2 patients have SOME insulin, so they rarely get DKA. Instead they get HHS (hyperosmolar hyperglycemic state). (so HHS, not DKA)
Obese patient, metabolic syndrome, older onset💡Type 2 = "Two heavy bags": metabolic syndrome + weight. The receptor has TWO problems: too much fat blocks the signal, and there are fewer receptors total (downregulation from chronic hyperinsulinemia).
Insulin receptor problem: the lock is jammed, the key is fine🔑HHS vs DKA: HHS = Hugely High Sugar (>600, osmolality >320, no ketones). DKA = Drowning in Ketoacid (pH <7.3, ketones 3+, glucose 250-500). The osmolality number is the board clue.
★The single fastest board question differentiator: DKA = Type 1. HHS = Type 2. Type 2 has enough insulin to prevent ketosis. Type 1 has zero.
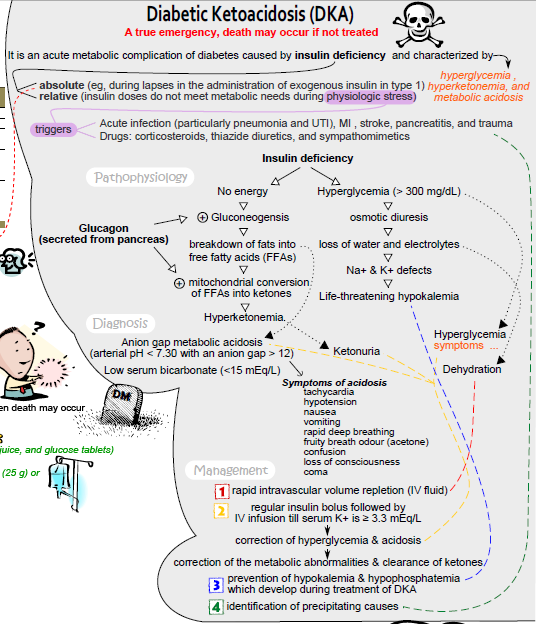
Concept map of DKA, where no insulin triggers lipolysis, ketoacid production, and Kussmaul breathing. Tap to expand.
Tap any cell to reveal. Test yourself first.
Feature
Type 1
Type 2
Trace It
Autoimmune beta cell destruction
Insulin resistance + relative deficiency
Antibody
Anti-GAD, anti-islet cell (ICA)
None
HLA
DR3, DR4
Not HLA-linked
Peak onset
Childhood (peak 3 to 4 years)
Adulthood (>40)
Body habitus
Thin, weight loss
Obese, metabolic syndrome
Acute crisis
DKA (fruity breath, pH <7.3)
HHS (extreme glucose >600, osmolality >320)
C-peptide
Low/absent (no insulin production)
Normal/high (insulin being made, not used)
Treatment
Insulin always
Metformin first, then add agents
CV mortality drugs
Insulin
SGLT2i + GLP-1 (the two that save lives)
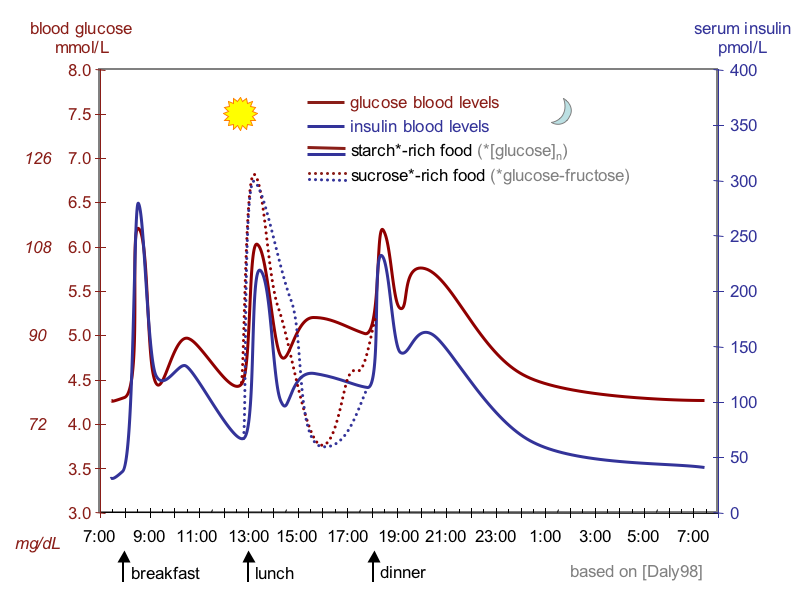
Daily glucose and insulin curves showing why C-peptide marks insulin the body made itself. Tap to expand.
⚠️
Board Trap: TZDs in Heart Failure
Thiazolidinediones (pioglitazone, rosiglitazone) are contraindicated in heart failure. They cause fluid retention and edema. The drugs that DECREASE cardiovascular mortality are SGLT2 inhibitors (-gliflozin) and GLP-1 agonists (-tide). Not TZDs.
SEE THE MECHANISM
The Insulin Key and the GLUT4 Door
Glucose can only cross into muscle and fat when insulin opens the GLUT4 door. Pick a scenario and watch what happens.
Why the same glucose ends up trapped or absorbed
In Type 1 there is no insulin key. In Type 2 the key arrives but the lock is jammed by resistance. Healthy cells open the door and glucose pours in.
Bloodstream
Inside the cell
🔑
Healthy cell: the insulin key docks, GLUT4 opens, and glucose moves inside.
FLIP CARDS
Know Each Type Cold
Tap a card to reveal the board-tested details for each presentation.
Tap any card to flip
💥
Type 1: The Attack
Pathophysiology & antibodies
Type 1 Pathophysiology
T-cell mediated autoimmune destruction of beta cells
Anti-GAD (anti-glutamic acid decarboxylase) most tested
Anti-islet cell antibodies (ICA-512) also positive
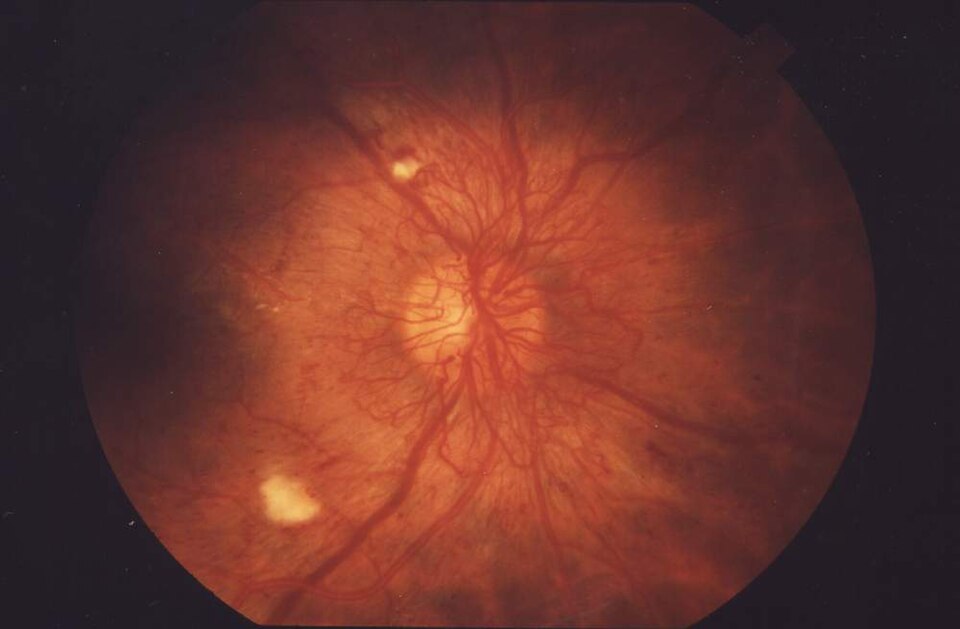
A fundus photo with neovascularization, the microvascular damage of chronic hyperglycemia. Tap to expand.
THE DRILL
BRICK-L: Organs That Never Needed Insulin
Seven tissues take up glucose without insulin. If you forget them, you will miss a board question.
Why This Matters
Most cells need insulin to move GLUT4 transporters to the surface so glucose can enter. But some tissues use GLUT1, GLUT2, or GLUT3GLUT1: brain, RBC (constitutive, always on). GLUT2: liver, kidney, intestine (bidirectional, no rate limit). GLUT3: neurons (high affinity). GLUT4: muscle, adipose (insulin-dependent). GLUT5: fructose transporter. instead, which are always active and do NOT require insulin signaling.
In a diabetic patient, these organs keep getting glucose even when insulin is absent or ineffective. This is why the brain still works in DKA (glucose gets in without insulin).
Tap All 7 BRICK-L Organs
Which of these organs do NOT need insulin for glucose uptake? Tap all 7. Wrong taps shake.
💡Brain, RBC, Intestinal wall, Cornea, Kidney, Liver, and eXercising muscle. BRICK-L (+ exercising muscle). The brain uses GLUT1/3, the liver uses GLUT2, and exercising muscle pulls GLUT4 to the surface through contraction alone.
⚠️
Board Trap: Resting vs Exercising Muscle
Resting skeletal muscle needs insulin (GLUT4 requires insulin signaling). Exercising muscle does NOT need insulin: muscle contraction alone translocates GLUT4 to the surface. This is why exercise lowers blood glucose even without insulin.
Nesidioblastosis
A baby born to an uncontrolled diabetic motherMaternal hyperglycemia crosses the placenta. The fetal pancreas responds by growing more beta cells (islet cell hyperplasia) to handle the extra glucose. After birth, the glucose supply stops, but the oversized pancreas keeps pumping out insulin, causing neonatal hypoglycemia. has overgrowth of the islet cells. After birth, those extra beta cells keep producing insulin even though the maternal glucose supply is gone.
Insulin: HIGH (too many beta cells)🧠Nesidioblastosis trick: "Nesi-BLAST-osis" = the pancreas was BLASTED with extra beta cells from maternal glucose. Baby is born, sugar supply cuts off, but those extra cells keep firing. High insulin + high C-peptide + low glucose = body made too much insulin on its own.
C-peptide: HIGH (endogenous insulin production confirmed)
Glucose: LOW (hyperinsulinemia drives glucose into cells)
This is the neonatal version of insulinoma: too much insulin, too little glucose.
CHALLENGE BEFORE REVEAL
Triage the Hyperglycemic Crisis
Commit to an answer first. The consequence unlocks only after you choose.
A teenager arrives obtunded with fruity breath, glucose 540 mg/dL, pH 7.10, and 3+ urine ketones. Which single value confirms the emergency you are treating?
DKA is defined by the acidosis, not the glucose. The high anion gap plus ketones is the diagnostic anchor; DKA glucose is usually only 250 to 500. Osmolality above 320 and glucose above 600 describe HHS, the Type 2 crisis. Break it down: acidosis plus ketones equals DKA; extreme osmolality equals HHS.
You start normal saline and an insulin drip. Two hours later glucose is 250 mg/dL and potassium has fallen from 5.6 to 3.1 mEq/L. What is the next move?
Insulin drives potassium into cells. The arrival potassium was falsely high from acidosis; total body stores were depleted. With a level of 3.1, hold insulin below 3.3 and replace potassium first, or you risk a fatal arrhythmia. Bicarbonate only worsens the hypokalemia. Break it down: replace potassium before you keep pushing insulin.
The patient stabilizes and is confirmed to have new Type 1 diabetes. Which long-term outpatient regimen is correct?
No beta cells, no oral agents. Type 1 has absolute insulin deficiency, so oral secretagogues have nothing to stimulate. Lifelong insulin is required, given as basal plus bolus to mimic the pancreas. Break it down: Type 1 always needs insulin; oral agents need living beta cells.
DECISION TREE
DM Management: Which Drug Next?
Work through the algorithm. Each branch reveals the board-tested rationale.
Start Here
New diagnosis of diabetes. Is the patient in acute crisis (glucose >600, altered mental status, or pH <7.3)?
Acute Crisis: Determine DKA vs HHS first. DKA (pH <7.3, ketones 3+, glucose 250-500) = Type 1 until proven otherwise. HHS (glucose >600, osmolality >320, no ketones) = Type 2 most likely. Both need IV fluids + electrolyte repletion. DKA requires insulin drip. HHS requires aggressive fluid resuscitation first.
Stable patient. Move to Step 2: determine diabetes type.
↓
Step 2: Type Differentiation
Is there evidence of autoimmune destruction? (positive anti-GAD, young/thin patient, DKA history, low C-peptide)
Type 1 DM: Insulin is required, always. No oral agents will work because there are no beta cells to stimulate. Start with basal-bolus insulin regimen (long-acting + rapid-acting with meals). Educate on carb counting, glucose monitoring, hypoglycemia recognition. Anti-GAD confirms the diagnosis.
Type 2 DM confirmed. Move to Step 3: first-line therapy.
↓
Step 3: First-Line T2DM
Does the patient have contraindications to metformin? (eGFR <30, contrast dye upcoming, acute illness)
Metformin alternatives: If eGFR 30-45, reduce dose. If eGFR <30, stop. Alternatives: GLP-1 agonist (preferred if obesity/CVD), SGLT2i (preferred if HF), DPP-4i (weight-neutral, renal-dose-safe). Sulfonylurea is last resort (hypoglycemia + weight gain).
Start metformin: 500mg daily with meals, titrate up over 4-8 weeks to 1000mg twice daily. Reduces hepatic glucose output. No hypoglycemia risk alone. No weight gain. First-line for nearly all T2DM. Move to Step 4 if A1c not at goal in 3 months.
↓
Step 4: Add-On Agents (A1c still above goal)
Which comorbidity matters most?
SGLT2 inhibitor: empagliflozin or dapagliflozin. Reduces CV mortality AND hospitalizations for HF. Mechanism: osmotic diuresis reduces preload + direct cardiac benefits. Avoid if eGFR <30. Side effects: genital mycotic infections, DKA risk (rare in T2).
GLP-1 agonist: semaglutide or liraglutide. Reduces MACE (major adverse cardiovascular events). Also causes significant weight loss (7-15%). Mechanism: incretin effect + delayed gastric emptying + central satiety. Injection or weekly oral (semaglutide). Avoid in medullary thyroid cancer history.
Flexible choices: Sulfonylurea (cheap, effective, hypoglycemia risk), DPP-4i (weight-neutral, expensive), or TZD (improves insulin sensitivity but contraindicated in HF). Pick based on cost, tolerability, and patient preference.
DECISION TREE
Decision Tree: Initial Diabetes Management
Pick a branch at each step. The rationale unlocks as you go.
What type of diabetes has been diagnosed?
Insulin is REQUIRED. No oral agents will stimulate beta cells that no longer exist. Start basal-bolus regimen: long-acting (glargine) nightly + rapid-acting (lispro or aspart) with meals. Teach carb counting. Add CGM. Target A1C below 7%.
Step 1: Lifestyle modification plus metformin first line (hold if eGFR is below 30). Recheck HbA1c in 3 months. If not at goal, add a second agent based on comorbidities (see branches below).
Add GLP-1 receptor agonist (semaglutide or liraglutide) OR SGLT2 inhibitor (empagliflozin). Both reduce major adverse cardiovascular events. GLP-1 agonists also drive significant weight loss. Either is acceptable; patient comorbidity profile guides the pick.
SGLT2 inhibitor preferred (empagliflozin or dapagliflozin). Reduces HF hospitalizations and slows CKD progression via hemodynamic and anti-inflammatory mechanisms. Avoid if eGFR is below 30. Watch for genital mycotic infections.
GLP-1 receptor agonist (semaglutide preferred for magnitude of weight loss). Mechanism: incretin effect, delayed gastric emptying, central satiety. Avoid in personal or family history of medullary thyroid carcinoma or MEN 2.
DPP-4 inhibitor (sitagliptin, linagliptin). Weight-neutral. Extremely low hypoglycemia risk because it only potentiates incretin effect in a glucose-dependent manner. Good renal dosing profile. No cardiovascular mortality benefit but very safe.
Sulfonylurea (glipizide or glimepiride) is cheap and effective. Mechanism: closes K-ATP channels on beta cells, forcing insulin release regardless of glucose level. Downside: hypoglycemia risk (especially if meal is skipped) and weight gain. Not the first choice if alternatives are affordable.
Consider early insulin regardless of type. At HbA1c above 10%, glucose toxicity itself impairs beta cell function. Temporary insulin can break the cycle, allow beta cell recovery, and sometimes be discontinued later once control is established. Reassess type and need after 3 months.
CHALLENGE
Clinical Questions
5 randomized from a pool of 10. Reload for a fresh set.
Medically reviewed by Kaitlyn Cocuzzo, MD and Fatima Ali, DO · Last updated July 2, 2026 at 3:42 PM ET
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.