One deletion on chromosome 7. The chloride channel fails. Five organ systems pay the price. Learn the mechanism chain that explains every manifestation, from salty sweat to bronchiectasis to infertility.
The Root Cause
Chromosome 7, Both Parents
Every board question on CF inheritance points to the same answer. Lock it in with the Punnett square and the X-linked trap before you see any stem.
A 4-year-old boy has recurrent Pseudomonas aeruginosa pneumonia, malodorous oily stools, and failure to thrive. Sweat chloride is 78 mEq/L. Both parents are healthy, non-consanguineous, and have no respiratory problems. The physician orders genetic testing on both parents. Which of the following findings would be expected?
What does genetic testing show in the parents?
Start with the gene. The CFTR geneCystic Fibrosis Transmembrane conductance Regulator. Encodes the ATP-gated chloride channel expressed in epithelial cells of the lungs, pancreas, sweat glands, and reproductive tract. sits on chromosome 7. The most common mutation is a three-base-pair deletion that removes a phenylalanine at position 508 of the CFTR protein, called delta-F508. This deletion causes the protein to misfold inside the endoplasmic reticulum, where it is degraded before it can reach the cell membrane.
Two copies, both broken. CF is autosomal recessive. To be affected, a child must inherit a defective CFTR allele from each parent. Carriers (one normal copy, one defective) are clinically healthy. The key board fact: both carrier parents have the mutation on chromosome 7, not the X chromosome.
Autosomal RecessiveTap to flip
Both chromosomes must carry the mutationAutosomal means the gene is on a non-sex chromosome. Recessive means one functional copy is enough to prevent disease. Both parents must each carry one defective CFTR allele on their chromosome 7 for a child to have CF. Unaffected carriers are heterozygous (Cc).
Chromosome 7 · delta-F508Tap to flip
The specific mutation, the specific chromosomeCFTR gene → chromosome 7. Delta-F508 mutation → phenylalanine deleted at position 508 → protein misfolds in the ER → degraded before reaching the cell surface → no functional Cl⁻ channel at the membrane.
NOT X-LinkedTap to flip
The classic inheritance trapX-linked recessive conditions (hemophilia, G6PD deficiency, Duchenne MD, chronic granulomatous disease) affect males almost exclusively. CF affects males and females equally because the gene is on chromosome 7, not X. Both sexes inherit chromosomes 1-22 in pairs. Sweat chloride plus equal sex distribution = autosomal, not X-linked.
CGD vs CF TrapTap to flip
Chronic granulomatous disease separates fastCGD (CYBB gene, X chromosome) causes recurrent catalase-positive bacterial infections (Staph, Aspergillus, Nocardia). CF causes recurrent Pseudomonas lung infections plus pancreatic insufficiency, salty sweat, and GI disease. If the stem has steatorrhea and a salty sweat test result: CF, autosomal recessive, chromosome 7. If it is a boy with recurrent Staph and Aspergillus abscesses: CGD, X-linked.
The carrier cross. When two carriers (each with one normal and one defective CFTR allele on chromosome 7) have children, the 2x2 Punnett square predicts:
C (normal)
c (mutant)
Father
CC 25% unaffected
Cc 25% carrier
Mother
Cc 25% carrier
cc 25% CF
Each pregnancy: 25% affected, 50% carrier, 25% unaffected. Inheritance is the same for sons and daughters.
Board Trap
A question stem with "recurrent bacterial infections in a child" can describe both CF and chronic granulomatous disease. The separating clues: CF adds salty sweat, steatorrhea, and pancreatic failure; CGD does not. CF is autosomal recessive on chromosome 7; CGD is X-linked recessive on the X chromosome. The sex of the patient and the sweat chloride result separate them every time.
From the Attending
When a board question asks about inheritance and the patient has CF, the answer is always autosomal recessive and chromosome 7. Not X-linked. Not chromosome X. The question sometimes buries a healthy carrier mother and a healthy carrier father in the history specifically to test whether you know the gene is autosomal. Chromosome 7. Both parents. Every time.
The Mechanism
What Happens When the Channel Fails
The delta-F508 protein misfolds and never reaches the cell surface. Toggle between normal and CF states to see exactly what changes and why thick mucus results.
Airway Lumen (above cells)
Thin, watery mucus → cilia clear it
Cl⁻
↑
out of cell
H₂O
↑
follows Cl⁻
CFTR OPEN ✓
Cell Cytoplasm
CFTR is properly folded → trafficked to membrane → ATP gates it open → Cl⁻ secreted into lumen → Na⁺ follows passively → water follows osmotically → thin mucus
Normal CFTR at the membrane → Cl⁻ secreted into airway lumenNa⁺ follows passively → water follows both ions osmoticallyMucus layer is well-hydrated → cilia beat freely and clear mucusBacteria are swept out before colonization → lungs stay clean
ΔF508 misfolds in the ER → degraded by proteasome → never reaches membrane. ENaC (epithelial Na⁺ channel) compensates by reabsorbing Na⁺ and water from lumen → dehydrated, sticky mucus
No CFTR at membrane → no Cl⁻ secretion → no osmotic gradient to pull water into lumenENaC hyperactive → Na⁺ (and water) reabsorbed OUT of lumen → mucus layer dehydratesThick mucus → cilia are immobilized → bacteria not cleared → Pseudomonas colonizesChronic infection → inflammation → airway wall destruction → bronchiectasis
Why salty sweat? CFTR in the sweat glandsIn the sweat duct, CFTR reabsorbs chloride from the sweat fluid back into the body. When CFTR is absent, chloride stays in the sweat. More chloride in sweat means the sweat tastes salty and the sweat chloride test result is elevated. works in the OPPOSITE direction from the lungs: it reabsorbs Cl⁻ from sweat back into the body. When CFTR fails, Cl⁻ stays in the sweat → sweat chloride concentration rises above 60 mEq/L → positive sweat chloride test. The affected child literally tastes salty when kissed, which is a classic clinical clue and the origin of the pilocarpine-induced sweat test.
From the Attending
The same CFTR protein does two opposite jobs depending on where you are. In the lungs and gut, CFTR secretes Cl⁻ into the lumen to hydrate secretions. In the sweat duct, it reabsorbs Cl⁻ to keep electrolytes in the body. Lose CFTR and you get thick mucus in the lungs AND salty sweat from the skin. One mechanism, two opposite-seeming consequences. Know both.
The Consequences
Every System Downstream of Mucus
Thick, sticky secretions obstruct every organ that depends on a thin mucus layer to function. Tap each system to reveal the mechanism chain and the board-testable findings.
Digital clubbing
CFTR channel (3D render)
Bronchiectasis CT
Meconium ileus X-ray
Lungs → Bronchiectasis
Tap to reveal mechanism
Thick mucus → impaired mucociliary clearance → bacteria colonize (first S. aureus in infancy, then mucoid Pseudomonas aeruginosa in adulthood) → recurrent infections → chronic inflammation → permanent airway dilation (bronchiectasis). Bronchiectasis appears on CT as dilated airways exceeding the diameter of their accompanying artery ("signet ring" sign). Digital clubbing develops from hypoxia and local growth factor release.
Pancreas → Insufficiency
Tap to reveal mechanism
Thick secretions block pancreatic ducts → digestive enzymes (lipase, amylase, proteases) cannot reach the small intestine → exocrine pancreatic insufficiency. Fat malabsorption causes steatorrhea (greasy, foul-smelling, floating stools) and deficiency of fat-soluble vitamins: A, D, E, K. Night blindness (Vit A), osteoporosis (Vit D), hemolytic anemia (Vit E), and bleeding (Vit K) follow. Immunoreactive trypsinogen is elevated on newborn screening because blocked ducts prevent trypsinogen from draining.
GI → Meconium Ileus
Tap to reveal mechanism
Thick, tenacious meconium obstructs the terminal ileum in the newborn. Presents as failure to pass meconium in the first 24-48 hours of life. Abdominal X-ray shows a "ground glass" appearance in the right lower quadrant (meconium mixed with bowel gas). Water-soluble contrast enema is both diagnostic and therapeutic. Meconium ileus is the presenting feature in 10-15% of CF newborns and is essentially pathognomonic for CF until proven otherwise.
Reproductive → Infertility
Tap to reveal mechanism
Males: congenital bilateral absence of the vas deferens (CBAVD). The vas deferens develops from the Wolffian duct, which requires CFTR-driven secretions to form properly. Without CFTR, the vas deferens does not form → obstructive azoospermia. Testes and spermatogenesis are normal; sperm simply cannot exit. This is the cause of infertility in over 97% of men with CF. Females are subfertile due to thick cervical mucus impairing sperm penetration.
Sweat Glands → Salty Sweat
Tap to reveal mechanism
CFTR in the sweat duct normally reabsorbs Cl⁻ back into the body. When absent, Cl⁻ stays in sweat → sweat chloride above 60 mEq/L → positive pilocarpine iontophoresis (sweat chloride test). This also causes a contraction alkalosis during heat stress: chronic Cl⁻ loss from skin → HCO₃⁻ retention compensates → hypochloremic metabolic alkalosis with hypokalemia.
Liver → Biliary Obstruction
Tap to reveal mechanism
Thick bile obstructs bile ducts → focal biliary cirrhosis in a subset of CF patients. Presents with portal hypertension, hepatomegaly, and eventually liver failure. Less clinically prominent than pulmonary and pancreatic disease but an important late complication in long-lived CF patients. Liver disease accounts for a significant portion of CF mortality after pulmonary disease.
From the Attending
Every CF complication traces back to the same two words: thick mucus. The lung traps bacteria. The pancreatic duct traps enzymes. The bile duct traps bile. The vas deferens never forms. The sweat duct loses chloride. If you understand that CFTR secretes fluid into lumens to keep things moving, the absence of CFTR explains everything. Thick secretions obstruct every lumen in the body. That is the whole disease.
Confirming It
Sweat Chloride, Newborn Screen, and the CFTR Modulators
Three diagnostic decision points and a comparison table that shows what each test actually measures and when to use it.
The sweat chloride test. Pilocarpine is applied to the skin via iontophoresis to stimulate sweat secretion. The sweat is collected and the chloride concentration is measured. Normal: below 40 mEq/L. Borderline: 40-59 mEq/L. Positive for CF: 60 mEq/L or above. The test must be performed twice on two separate occasions. A single positive result is not sufficient for diagnosis. False negatives are rare but can occur in edematous newborns.
Newborn screening. Elevated immunoreactive trypsinogen (IRT)Trypsinogen is a pancreatic enzyme precursor. Normally it drains through the pancreatic duct into the intestine. When CF blocks the duct, trypsinogen backs up into the bloodstream, raising serum IRT. This is the newborn blood spot screen for CF. is the initial newborn screen marker. A positive IRT is followed by CFTR mutation analysis from the same blood spot. If two CFTR mutations are identified, the diagnosis is confirmed. If the result is ambiguous, a sweat chloride test is performed at 2 weeks of age or later.
A 3-week-old male infant fails to pass meconium and has elevated immunoreactive trypsinogen on the newborn screen. His parents are first-generation immigrants with no family history of CF. Which of the following best represents the correct next diagnostic step?
Elevated IRT on newborn screen is the trigger; the sweat chloride test is the confirmatory step. A chloride concentration above 60 mEq/L on two separate tests diagnoses CF. Sputum culture is irrelevant at 3 weeks before any pulmonary disease develops. Immunodeficiency panels address different conditions. Bronchiectasis takes years of repeated infection to develop. Elevated IRT in a newborn: next step is sweat chloride test.
The elevated IRT is the screen; the sweat chloride test (pilocarpine iontophoresis) is the confirmation. Sputum, imaging, and immune panels address other diagnoses. Sweat chloride greater than 60 mEq/L on two occasions confirms CF.
A 7-year-old girl with CF and known pancreatic exocrine insufficiency is found to have a prothrombin time of 24 seconds (INR 2.1). She is not on anticoagulants. Her diet has been poor for months due to nausea. Which fat-soluble vitamin deficiency most directly explains the coagulation abnormality?
The fat-soluble vitamins A, D, E, and K are all malabsorbed in CF due to pancreatic lipase insufficiency. Vitamin K is required for gamma-carboxylation of clotting factors II, VII, IX, and X. When Vitamin K is absent, the PT/INR rises because the extrinsic pathway (Factor VII) and common pathway (II, X) cannot be activated normally. The prolonged PT in a CF patient with poor nutrition and no anticoagulants is Vitamin K deficiency until proven otherwise. CF plus fat malabsorption plus prolonged PT: Vitamin K deficiency. Replace ADEK supplementation and add pancreatic enzymes.
The PT/INR tests the extrinsic and common coagulation pathways. Those factors (II, VII, IX, X) require Vitamin K for activation. Fat malabsorption in CF causes Vitamin K deficiency, raising the PT. A, D, and E have different manifestations.
A 19-year-old woman with CF presents with a 3-week exacerbation of cough, increased dyspnea, and decreased FEV1. Sputum culture grows mucoid Pseudomonas aeruginosa resistant to ciprofloxacin. She is admitted for inpatient treatment. Which antibiotic combination is the standard treatment backbone for a CF pulmonary exacerbation with Pseudomonas?
CF pulmonary exacerbations with Pseudomonas require two-drug anti-pseudomonal therapy to minimize resistance emergence. An anti-pseudomonal beta-lactam (ceftazidime, cefepime, piperacillin-tazobactam, meropenem) combined with an aminoglycoside (tobramycin, in particular, because inhaled tobramycin is a standard maintenance therapy) is the backbone. Vancomycin addresses MRSA, not Pseudomonas. Azithromycin has anti-inflammatory properties in CF but is not sufficient to treat an acute Pseudomonas exacerbation. Mucoid Pseudomonas in CF exacerbation: double coverage with anti-pseudomonal beta-lactam plus aminoglycoside.
Pseudomonas requires anti-pseudomonal coverage. Vancomycin targets gram-positive organisms. TMP-SMX covers MRSA. Azithromycin is maintenance, not acute treatment. Anti-pseudomonal beta-lactam plus aminoglycoside is the standard CF exacerbation regimen.
CFTR Modulators. New disease-modifying drugs target the underlying protein defect. Ivacaftor (a potentiator) keeps the channel open longer for gating mutations. Lumacaftor/tezacaftor (correctors) help the misfolded delta-F508 protein traffic to the membrane. Elexacaftor-tezacaftor-ivacaftor (Trikafta) is the triple combination that works for most patients with at least one delta-F508 allele and has dramatically improved survival. These drugs do not cure CF but substantially slow lung function decline.
Six original 3rd-order vignettes, 5 dealt per round, answer choices shuffled each time. Tap a wrong answer to see why it almost works, then read the glowing clues. Right-click or long-press to cross out distractors. Double-tap to highlight a choice.
Medically reviewed by Kaitlyn Cocuzzo, MD and Fatima Ali, DO · Last updated July 1, 2026 at 10:03 PM ET
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.
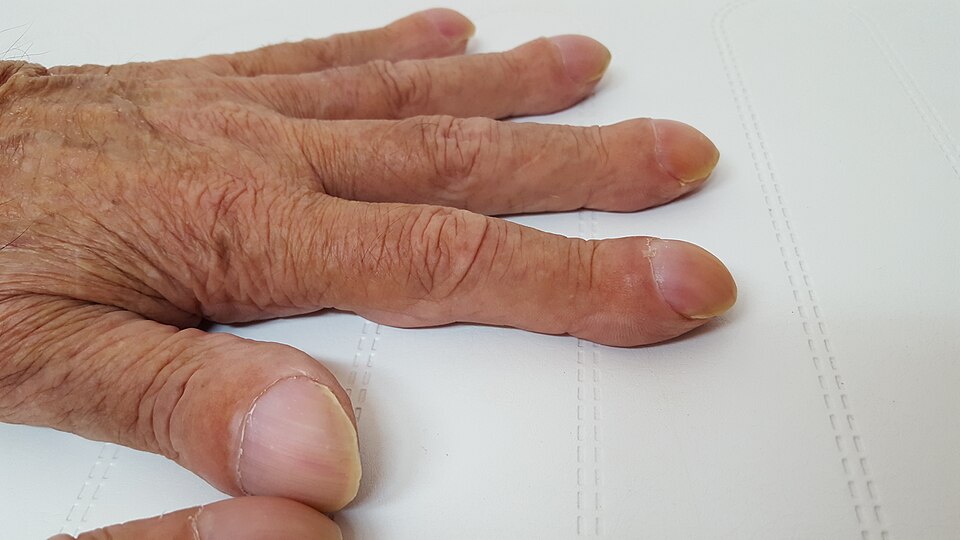 Digital clubbing
Digital clubbing
 CFTR channel (3D render)
CFTR channel (3D render)
 Bronchiectasis CT
Bronchiectasis CT
 Meconium ileus X-ray
Meconium ileus X-ray