Why a narrowed kidney artery turns into whole-body hypertension -- and why it won't respond to your drugs.
A 64-year-old man with a history of smoking and peripheral vascular disease presents with resistant hypertension despite maximal doses of three antihypertensives. Renal angiography reveals 90% stenosis of the right renal artery. A biopsy of the juxtaglomerular apparatus would most likely show:
A. Efferent arteriole dilation
B. Juxtaglomerular cell hyperplasia
C. Decreased renin secretion
D. Macula densa atrophy
E. Mesangial cell proliferation
From the Attending
Resistant hypertension in an older smoker with vascular disease is not bad luck. It is a clogged renal artery talking. The kidney behind that clog cannot tell the difference between a stenosis and true shock, so it screams for pressure the only way it knows how: renin. One narrowed pipe, a whole body under pressure. Find the source.
THE PATTERN
One Clogged Pipe, Whole-Body Chaos
Why a problem in ONE kidney artery raises blood pressure EVERYWHERE
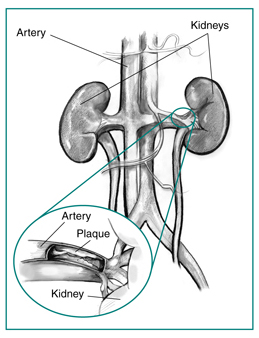
Here's the setup. Your kidney doesn't just filter blood · it monitors blood pressure. There's a cluster of specialized cells wrapping around the afferent arterioleThe vessel that brings blood INTO the glomerulus. "Afferent" = Arriving. The JGA cells sit here like bouncers checking the pressure before letting blood through. called the juxtaglomerular apparatus (JGA)A pressure-sensing station where the afferent arteriole meets the distal convoluted tubule. Contains JG cells (sense pressure) and macula densa (sense sodium). Together they control renin release.. Their one job: sense pressure.
When that renal artery gets squeezed to 90% closed, the kidney downstream sees drastically less blood flow. The JGA's pressure sensors scream: "We're losing perfusion!" Their response? Dump renin.
🔑Renal Artery Stenosis = Renin Always Spiking
Renin is the match that lights the whole RAAS fire. And when the stenosis is permanent, the match never goes out. The JGA cells don't just work harder · they multiply. That's JGA hyperplasia: more cells making more renin, day after day, month after month.
The kidney thinks it's in shock. It's not · it's just downstream of a clog. But it can't tell the difference. So it keeps screaming for more pressure, forever.
The logic chain
1
A stenosis is a local plumbing problem. The pressure sensor at the JGA only knows local pressure, not the reason it dropped.
2
Low pressure at the afferent arteriole reads as "the whole body is underperfused," so the JGA dumps renin into the systemic circulation.
3
Renin raises pressure everywhere, including in the healthy kidney on the other side. That kidney is now hypertensive but cannot turn the renin off, because the off-switch lives behind the stenosis.
4
The stenosis never resolves on its own, so the signal never stops. Chronic stimulation makes the JG cells proliferate: that is JG hyperplasia on biopsy.
One kidney's local low-pressure reading becomes the whole body's hypertension, and the only kidney that could shut it off is the one that cannot sense the high pressure.
From the Attending
Do not call this essential hypertension. Essential hypertension does not give you an abdominal bruit and it does not start in a 30-year-old woman. When the renin is high and the pressure will not come down on three drugs, the kidney is the engine. High renin driving high pressure is secondary hypertension until you prove otherwise.
THE CASCADE
RAAS: The Domino Chain
Fire it as one sequence, then walk each step below
The pressure alarm
One Pinched Pipe
A stenosis throttles flow to one kidney. Its pressure sensor reads a drought and arms the whole cascade.
Renal perfusion: low · MAP 112
Pattern locked
Renin to Pressure
RouteStenosis → JGA renin → ANG I (liver) → ANG II (lung ACE) → vessels + adrenal
PatternVasoconstriction plus aldosterone-driven Na and water retention: pressure up everywhere
PearlThe off-switch sits behind the stenosis, so the alarm never stops. High renin, high aldosterone.
🧊
Renal artery stenosis → decreased perfusion pressure at the afferent arteriole. JGA cells detect low stretch.
↓
R
Renin released from JG cells into the bloodstream. This is the rate-limiting step · everything downstream depends on this.
↓
L
Angiotensinogen (made by the liver, always floating around) gets cleaved by renin → Angiotensin IRENIN
↓
🫁
ACE (in lung endothelium) converts Angiotensin I → Angiotensin IIACE ACE also degrades bradykinin (vasodilator) · that's why ACEi cause cough.
↓
💥
Angiotensin II · the most powerful vasoconstrictor your body makes. Constricts efferent > afferent arteriole. Raises BP systemically.
↓
A
Aldosterone released from adrenal zona glomerulosa → Na+/H2O reabsorption in collecting duct. More volume = more pressure.
↓
🧠
ADH (vasopressin) release stimulated + thirst center activated. Even more water retention. The whole system is screaming: RAISE THE PRESSURE.
THE BOSS
Angiotensin II: 6 Ways It Wrecks You
Tap each card to see the mechanism
⭕ Vasoconstriction
Constricts arterioles body-wide. Efferent > afferent in the kidney · this preserves GFR even when perfusion drops.
Think of it like pinching the drain hose harder than the supply hose. Less blood gets out of the glomerulus, so filtration pressure stays up. That's why ACEi can DROP GFR in bilateral RAS · you're removing the only thing keeping filtration going.
🧂 Aldosterone Release
Stimulates zona glomerulosa → aldosterone → Na+ reabsorption + K+ secretion in the collecting duct.
More sodium reabsorbed = more water follows = more blood volume = higher BP. Also causes hypokalemia. Prolonged activation = metabolic alkalosis from H+ loss.
💧 ADH Secretion
Stimulates posterior pituitary to release ADH (vasopressin) → free water reabsorption via aquaporin-2 in collecting duct.
Double whammy: aldosterone grabs sodium, ADH grabs water. The body is pulling every lever to expand volume and raise pressure.
🤔 Thirst
Acts on the subfornical organ and OVLT in the hypothalamus → you feel thirsty → you drink more → volume goes up.
Your brain is literally conspiring with your kidneys to raise your blood pressure. The thirst signal is Ang II reaching the brain through circumventricular organs that lack a blood-brain barrier.
🔄 PCT Na Reabsorption
Stimulates the Na+/H+ exchanger in the proximal convoluted tubule. Grabs sodium early, before the loop even sees it.
This is the sneaky one. Aldosterone works on the collecting duct, but Ang II works on the PCT · hitting sodium reabsorption at two different sites along the nephron.
❤️ Cardiac Remodeling
Promotes hypertrophy and fibrosis in the heart and blood vessels. Long-term exposure → stiff, thick-walled ventricle.
This is why chronic RAAS activation is so destructive. It's not just the blood pressure · Ang II directly damages organs even independent of BP. ARBs and ACEi reduce this remodeling.
From the Attending
Angiotensin II is not just a vasoconstrictor. It grabs sodium at the proximal tubule, it pulls aldosterone out of the adrenal, it makes you thirsty, and it scars the heart. That is why we block this axis in heart failure and diabetic kidney disease, not only in hypertension. One hormone, sodium plus pressure plus remodeling. Block the axis, protect the organ.
THE BACKUP SENSOR
Tubuloglomerular Feedback
The second way your kidney detects low flow
The JGA has a buddy system. While JG cells sense pressure, the macula densaSpecialized cells in the wall of the thick ascending limb / early DCT, right where it touches the afferent arteriole. They sense NaCl concentration in the tubular fluid. Low NaCl = low GFR = release prostaglandins to trigger renin. senses sodium concentration in the tubular fluid.
1
Renal artery stenosis → less blood filtered → less NaCl delivered to the distal tubule
Prostaglandins signal JG cells → renin release (same endpoint, different trigger)
4
Prostaglandins also dilate the afferent arteriole → trying to increase GFR locally
This is why NSAIDs worsen renal function · they block prostaglandins, removing the kidney's backup rescue mechanism.
ACEi in bilateral RAS: Both kidneys depend on Ang II constricting the efferent arteriole to maintain GFR. Give an ACEi → efferent dilates → filtration pressure collapses → acute kidney injury. Board question: "New creatinine spike after starting lisinopril" = think bilateral RAS.
"Resistant hypertension" in clinical practice = hypertension despite 3+ drugs including a diuretic at max dose. Top 3 causes: renovascular disease (RAS), primary aldosteronism, pheochromocytoma. If the stem mentions smoking + PVD + resistant HTN, it's RAS until proven otherwise.
Two causes of RAS with different demographics: Atherosclerosis (older male, smoker, PVD) affects the proximal 1/3 of the renal artery. Fibromuscular dysplasia (young woman) affects the middle-to-distal renal artery · has a "string of beads" appearance on angiography.
🔑Atherosclerosis = Aortic end (proximal). FMD = Far out (distal).
See it on imaging · tap any image to expand
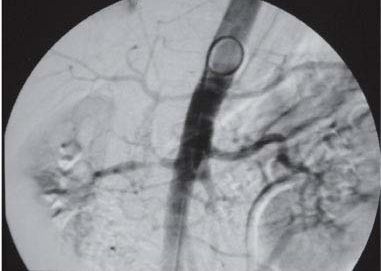
Atherosclerotic RASPlaque at the artery origin
Catheter angiogramThe gold-standard look
FMD: string of beadsArrows mark the beading
FMD: selective viewMid-to-distal artery
Atherosclerotic RAS vs Fibromuscular Dysplasia
Tap a cell to reveal. Test yourself before you peek.
Feature
Atherosclerotic
Fibromuscular dysplasia
Who
Older man, smoker, known vascular disease
Young woman, often under 50, otherwise healthy
Where in the artery
Proximal third, at or near the ostium by the aorta
Mid to distal artery, may extend into branches
Angiogram look
Single smooth focal narrowing, plaque shadow
String of beads: alternating stenosis and dilation
Angioplasty (usually no stent) works well, often cures
DECISION TREE
Diagnose It, Then Treat It
Guess first, then check yourself
The creatinine just jumped
You start lisinopril for blood pressure. Two weeks later the creatinine climbs from 1.1 to 3.0 with no protein and no casts. Before you read on: what does that single move tell you?
Bilateral disease. Both kidneys were leaning on angiotensin II to clamp the efferent arteriole and hold up filtration. Pull the angiotensin II with an ACE inhibitor and both efferents dilate at once, so filtration pressure collapses and the creatinine spikes. A unilateral stenosis usually tolerates an ACE inhibitor because the healthy kidney carries the load. A big creatinine jump right after starting an ACE inhibitor screams bilateral RAS or a single working kidney. Stop the drug, the creatinine comes back.
How you confirm it and what you do
1
Screen non-invasively first. Duplex ultrasound, CT angiography, or MR angiography show the narrowing without dye load to the kidney. Catheter angiography is the gold standard but is invasive, so it is saved for when you plan to intervene.
2
The captopril challenge is a stress test for the kidney. The stenotic kidney survives on angiotensin II holding its efferent arteriole tight. Give captopril, remove that angiotensin II, and that kidney's filtration drops, so its tracer uptake on a renogram falls behind the normal side. A side that wilts when you block the axis was living on it.
3
Most patients are managed medically. Control pressure and protect vessels: the large trials showed routine stenting of atherosclerotic lesions did not beat good medical therapy. Reach for revascularization when there is flash pulmonary edema, hypertension you cannot control on full therapy, or kidney function that is sliding.
4
Fibromuscular dysplasia is the exception that loves a fix. A young woman with mid-artery beading often gets a durable cure or a big drug reduction from angioplasty, usually without a stent.
ACE inhibitors and ARBs are first-line for one-sided disease and for the systemic RAAS, but they are the wrong move in bilateral RAS or a solitary stenotic kidney, where they can crash the only filtration the patient has.
RAAS Drugs: Pick Your Target
Pick your goal, follow the branch.
Goal of intervention?
ACE inhibitor (lisinopril, enalapril): reduces AII + bradykinin accumulates (cough, angioedema risk). Use in HFrEF, DM nephropathy, post-MI. Avoid in pregnancy and bilateral RAS.
ARB (losartan, valsartan): same effect as ACE-I, no bradykinin accumulation (no cough). Use if ACE-I intolerant. Avoid in pregnancy and bilateral RAS.
ARNI (sacubitril/valsartan): blocks neprilysin (raises natriuretic peptides) + blocks AT1 receptor. Superior to ACE-I in HFrEF. Do not combine with ACE-I (angioedema risk).
Spironolactone or eplerenone: block mineralocorticoid receptor. K-sparing. Use in HFrEF, primary hyperaldosteronism, cirrhosis. Risk: hyperkalemia, gynecomastia (spiro only).
Beta-blockers (metoprolol, carvedilol): block beta-1 receptors on JG cells, suppressing renin secretion. Use in HTN with tachycardia or HFrEF.
BOARD PRACTICE
Prove You Know the Cascade
5 patients. Their kidneys are mad. Figure out why.
Medically reviewed by Kaitlyn Cocuzzo, MD and Fatima Ali, DO · Last updated July 5, 2026 at 8:17 PM ET
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.
BOARD-STYLE WALKTHROUGH
Test Yourself
Six original board-style vignettes, one at a time. Right-click or long-press a choice to cross it out, double-tap to highlight. After you answer, tap the chain to walk the logic.