The enzymes that copy and proofread your genome, and the cancers and syndromes that happen when they fail.
Opening Challenge
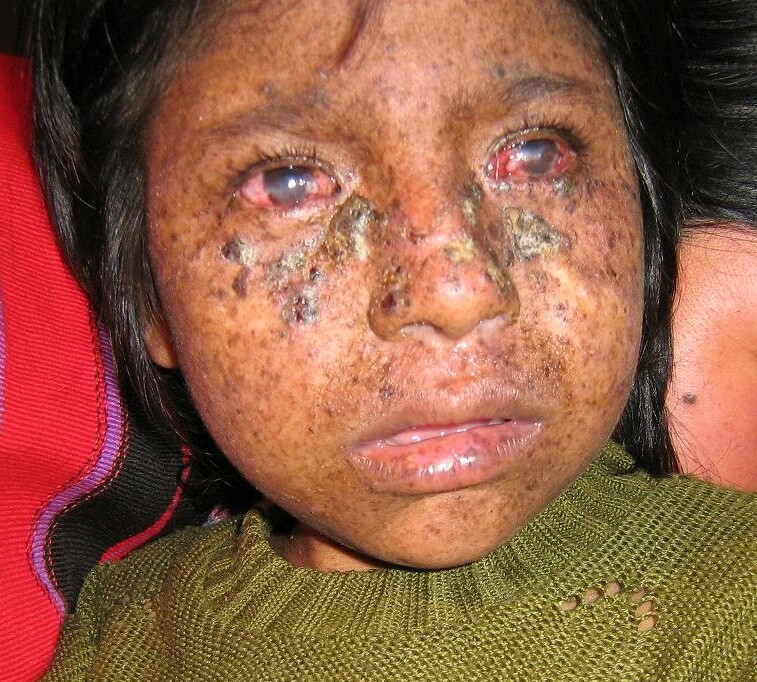
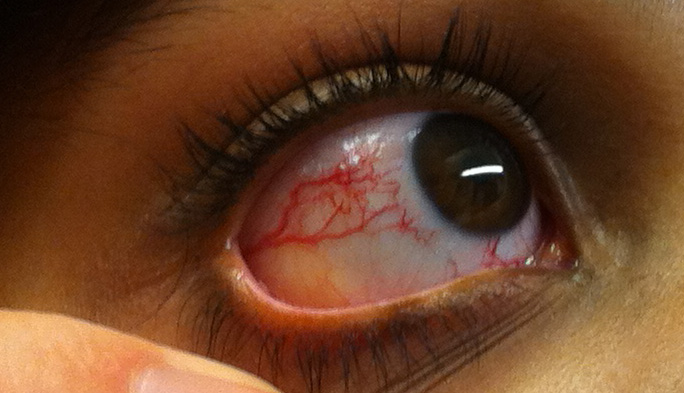
An 8-year-old boy is brought in by his parents after developing multiple blistering sunburns from brief outdoor exposure. On exam you find three basal cell carcinomas on his face and forearms. His parents report he has always been unusually sensitive to sunlight. Ophthalmology notes corneal opacities from UV exposure. Neurologic exam is normal.
A) Fanconi Anemia (crosslink repair defect)
B) Lynch Syndrome (mismatch repair defect)
C) Xeroderma Pigmentosum (nucleotide excision repair defect)
Xeroderma Pigmentosum (XP). UV light creates bulky pyrimidine dimers (thymine-thymine crosslinks on the same strand). The only pathway that removes these is Nucleotide Excision Repair (NER). XP patients carry mutations in any of 8 XP genes required for NER. Without NER, UV damage accumulates with every sun exposure, causing skin cancers at a very young age.
A (Fanconi Anemia): Good instinct on rare inherited repair syndromes, because Fanconi also involves DNA repair and causes cancer. But ask what damage Fanconi fixes: interstrand crosslinks that tie the two strands together, resolved through homologous recombination. Did UV light fuse the two strands together, or weld two pyrimidines on one strand into a bulky kink? The latter. And which crew handles a bulky single-strand kink? NER. The sun-sensitive child with childhood skin cancers points to the UV crew, not the crosslink crew. Fanconi anemia repairs interstrand crosslinks; UV pyrimidine dimers are bulky single-strand lesions cleared by NER, and losing NER is xeroderma pigmentosum.
B (Lynch Syndrome): Tempting because Lynch is a high-yield inherited cancer syndrome. But which damage does mismatch repair correct? Replication mismatches and slippage loops, and its failure targets colon and endometrium with microsatellite instability. Is a sun-sensitive child with skin cancer a colon-and-endometrium problem? No. The UV history and corneal clouding point to a bulky photoproduct, the NER substrate. Lynch is mismatch repair failure causing colorectal and endometrial cancer; UV-triggered skin cancer in a child is NER failure (xeroderma pigmentosum).
D (BRCA1): Ask what lesion BRCA1 handles: double-strand breaks, repaired by homologous recombination in dividing cells. Does ultraviolet light snap both strands across from each other, or distort one strand with a bulky dimer? The dimer is a single-strand bulky lesion, and the crew built for that is NER, not the double-strand-break crew. UV sensitivity with early skin cancers always routes back to NER. BRCA1 runs homologous recombination for double-strand breaks; bulky UV dimers are repaired by NER, and losing NER is xeroderma pigmentosum.
01 · Replication Machine
The Replication Machine
Three phases. Dozens of enzymes. The board tests initiation, fidelity, and how prokaryotes differ from eukaryotes.
Phase 1 · Starting the Machine
Initiation
Replication starts at specific DNA sequences called origins of replication (AT-rich in prokaryotes; multiple origins in eukaryotes for speed). Proteins recognize and unwind DNA here.
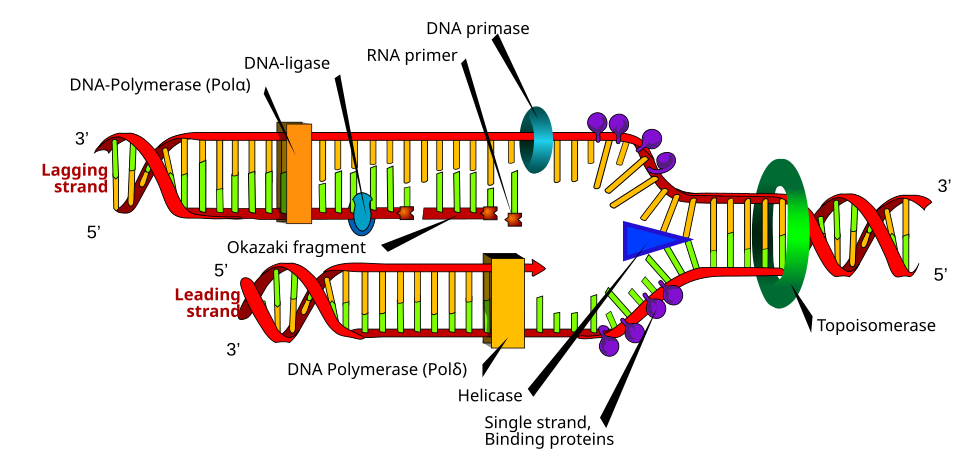
Helicase unwinds the double helix by breaking hydrogen bonds between base pairs, creating a replication fork. It moves in the 3' to 5' direction along the template strand, powered by ATP.
Single-strand binding proteins (RPA in eukaryotes, SSB in prokaryotes) coat the exposed single-stranded DNA to prevent re-annealing and protect it from nucleases.
Primase (an RNA polymerase) lays down a short RNA primer (~10 nucleotides). DNA polymerase cannot start a new chain from scratch and can only extend a pre-existing 3' OH. The primer provides that 3' OH.
AT-rich originsHelicase unwindsRPA/SSB stabilizesPrimase adds RNA primerDNA Pol needs 3'-OH to start
Origin recognition→Helicase unwinds→RPA coats ssDNA→Primase: RNA primer→DNA Pol extends
Why an RNA primer? DNA polymerase can only ADD nucleotides to a free 3' OH. It cannot start a chain on its own. Primase synthesizes a short RNA chain to provide that 3' OH. The primer is later removed and replaced with DNA.
Phase 2 · Copying the Strand
Elongation
DNA polymerase synthesizes new DNA 5' to 3' only. Since the two template strands run antiparallel, one strand is copied smoothly (leading strand) and the other in fragments (lagging strand).
Leading strand: synthesized continuously 5' to 3' toward the replication fork. One primer, one long chain.
Lagging strand: synthesized in short Okazaki fragments (1,000-2,000 nt in prokaryotes, 100-200 nt in eukaryotes). Each fragment needs its own RNA primer. The fragments go away from the fork, each 5' to 3'.
Prokaryote enzymes: DNA Pol III extends. DNA Pol I removes RNA primers (5' to 3' exonuclease) and fills the gap (5' to 3' polymerase). DNA ligase seals the final nick.
Eukaryote enzymes: DNA Pol alpha lays the primer + a short extension. DNA Pol delta (lagging strand) and DNA Pol epsilon (leading strand) extend the chains. RNase H removes RNA primers. Flap endonuclease (FEN1) helps. DNA ligase I seals nicks.
Leading: continuousLagging: Okazaki fragmentsPol III (prok): extendsPol I (prok): removes primersPol delta/epsilon (euk): extendsLigase seals nicks
The prokaryote vs. eukaryote split: Prokaryote: DNA Pol III extends, Pol I removes primers. Eukaryote: Pol alpha lays primer, Pol delta (lagging) and Pol epsilon (leading) extend. Remember "alpha starts, delta/epsilon finish."
Phase 3 · Quality Control
Fidelity & Chromosome Ends
3' to 5' exonuclease proofreading: Built directly into DNA polymerase. After inserting each nucleotide, the polymerase checks the most recent base pair. If wrong, the 3' to 5' exonuclease activity chews back and removes the mismatched nucleotide. Error rate drops from 1 in 10,000 to 1 in 10 million.
Mismatch repair (MMR): A separate repair system catches errors that slip past proofreading. MLH1, MSH2, MSH6, PMS2 recognize mismatch bulges after replication, excise the incorrect strand, and fill it in correctly.
Telomeres and telomerase: Each replication cycle, the lagging strand cannot fully copy the very end of a linear chromosome (the "end-replication problem"). This means chromosomes get shorter with each division. Telomeres (TTAGGG repeats) cap chromosome ends to protect coding sequences.
Telomerase is a reverse transcriptase (carries its own RNA template) that adds TTAGGG repeats back to chromosome ends. It is active in: stem cells, germline cells, and most cancer cells. It is inactive in: normal somatic cells. This is why somatic cells age and cancer cells don't.
3' to 5' proofreading in DNA PolMMR: catches post-replication errorsTelomeres: TTAGGG repeatsTelomerase: reverse transcriptaseActive in stem cells + cancerInactive in somatic cells (aging)
Board trap on telomerase: The question often asks "why is telomerase active in cancer cells?" Answer: cancer cells reactivate telomerase to prevent chromosomal shortening and achieve replicative immortality. Normal somatic cells have silenced telomerase, so they eventually senesce.
02 · Damage Control
Five Repair Pathways
Each pathway fixes a specific type of damage. The type of lesion tells you which pathway. The pathway tells you the disease.
DNA Damage Dispatch
Select a damage type to deploy the repair crew
FIXES:
KEY PROTEINS:
IF LOST:
The core pattern: damage type determines which repair pathway is used. When that pathway fails, a specific type of mutation accumulates, causing a characteristic cancer or syndrome. Every pathway below follows this logic.
01Nucleotide Excision Repair (NER)
Bulky lesions▸
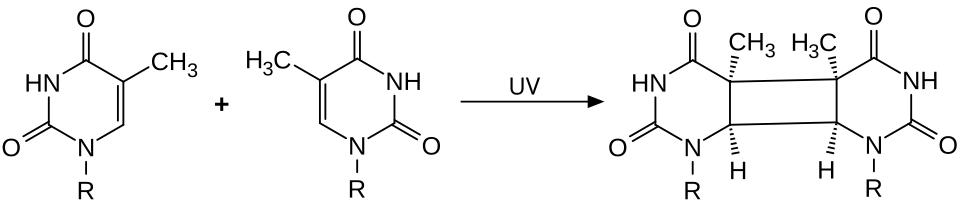
What it fixes: Bulky helix-distorting lesions: UV-induced pyrimidine dimers (thymine-thymine cyclobutane dimers and 6-4 photoproducts), chemical adducts from carcinogens like benzo[a]pyrene.
Mechanism: A multi-protein complex recognizes the helix distortion. Endonucleases cut the damaged strand on both sides, removing an oligonucleotide ~25-30 nucleotides long. DNA polymerase fills the gap using the opposite strand as template. Ligase seals it.
Transcription-coupled NER (TC-NER): A specialized sub-pathway that operates on actively transcribed genes. When RNA polymerase stalls at a lesion, TC-NER factors (CSA, CSB) are recruited to preferentially repair that site. This is why transcription-coupled NER defects cause a different syndrome than global NER defects.
Diseases:
- Xeroderma Pigmentosum (XP): mutations in any of 8 genes (XPA through XPG, plus XPV). Global NER is lost. UV skin cancers, extreme sun sensitivity, neurodegeneration in some forms.
- Cockayne Syndrome: mutation in CSA or CSB (TC-NER only). Progeria-like accelerated aging, photosensitivity, NO increased cancer risk (because global NER is intact, just transcription-coupled is lost).
Mnemonic: "NER = Nobody Escapes Radiation damage if you have XP." XP loses global NER; Cockayne loses TC-NER only.
02Base Excision Repair (BER)
Small base damage▸
What it fixes: Small, non-helix-distorting base modifications: deamination (cytosine converted to uracil), oxidation (8-oxoguanine from reactive oxygen species), alkylation, and some abasic sites.
Mechanism:
1. DNA glycosylase recognizes and flips out the damaged base, cleaves the glycosidic bond, leaving an AP (abasic/apurinic/apyrimidinic) site.
2. AP endonuclease (APE1) cuts the phosphodiester backbone at the AP site.
3. DNA Pol beta fills in the 1-nucleotide gap (short-patch BER).
4. Ligase III + XRCC1 seals the nick.
Classic example: Deamination of cytosine to uracil occurs ~500 times/day/cell. Uracil-DNA glycosylase (UDG) removes the uracil before it causes a C to T transition mutation.
Disease: MUTYH-associated polyposis. MUTYH is a DNA glycosylase that removes adenine when it is mismatched opposite 8-oxoguanine (an oxidized guanine). Without MUTYH, G:C to T:A transversions accumulate, driving colon polyps. Autosomal recessive, unlike APC-driven FAP.
Mnemonic: BER = "Base Erasing Robot." Glycosylase erases the base first, then the rest fills in.
03Mismatch Repair (MMR)
Replication errors▸
What it fixes: Replication errors that slip past proofreading: base-base mismatches (wrong base inserted) and insertion/deletion loops (IDLs) at repetitive sequences. IDLs cause microsatellite instability (MSI) when MMR fails.
Mechanism: MSH2-MSH6 (MutS homolog complex) recognizes the mismatch bulge. MLH1-PMS2 (MutL homolog complex) is then recruited. Together they mark the newly synthesized strand (which contains the error), excise a stretch of DNA around the mismatch, and recruit Pol delta to fill it in. Ligase seals it.
Key proteins: MLH1, MSH2, MSH6, PMS2 (the four Lynch syndrome genes).
Disease: Lynch Syndrome (HNPCC, Hereditary Non-Polyposis Colorectal Cancer). Autosomal dominant. Mutations in MLH1, MSH2, MSH6, or PMS2. The most common hereditary colorectal cancer syndrome. Also causes endometrial, ovarian, gastric, and urinary tract cancers.
Testing: Microsatellite instability (MSI-H) on tumor testing. Immunohistochemistry (IHC) for MMR protein expression loss. Germline testing confirms the gene.
Mnemonic: "MMR = Mistakes Must be Removed." Lynch = Loss of MMR = Lots of Microsatellite instability.
04Non-Homologous End Joining (NHEJ)
Double-strand breaks▸
What it fixes: Double-strand breaks (DSBs) in G1 phase (when no sister chromatid is available as template). Also used for V(D)J recombination to generate antibody and T-cell receptor diversity.
Mechanism: Error-prone because it does NOT use a template. Ku70/Ku80 heterodimer binds the broken ends and recruits DNA-PKcs (DNA-dependent protein kinase catalytic subunit). The ends are processed (trimmed), then ligated directly by XRCC4-Ligase IV complex. No template = potential for small insertions or deletions at the join site.
Disease: Severe combined immunodeficiency (SCID). V(D)J recombination during lymphocyte development requires NHEJ to join the cut gene segments. Defects in Artemis (Artemis-SCID), DNA-PKcs, or Ligase IV abolish V(D)J recombination, leaving T and B cells non-functional. Also: radiosensitivity syndromes.
Mnemonic: NHEJ = "Not Here, Errors Join." G1 phase, no template, joins are messy. V(D)J relies on this messiness to create diversity.
05Homologous Recombination (HR)
Error-free DSB repair▸
What it fixes: Double-strand breaks (DSBs) in S and G2 phases (when the sister chromatid is available as a high-fidelity template). Also repairs interstrand crosslinks (ICLs). Error-free.
Mechanism: After a DSB, the broken ends are resected to create 3' single-strand overhangs. BRCA2 loads RAD51 onto these 3' tails. RAD51 filaments catalyze strand invasion into the intact sister chromatid. The intact sequence is used as a template to rebuild the broken region with perfect fidelity. Resolution of the Holliday junction completes repair.
Key proteins: BRCA1 (coordinates end resection and signaling), BRCA2 (loads RAD51), RAD51 (strand invasion), RPA (coats initial ssDNA).
Diseases:
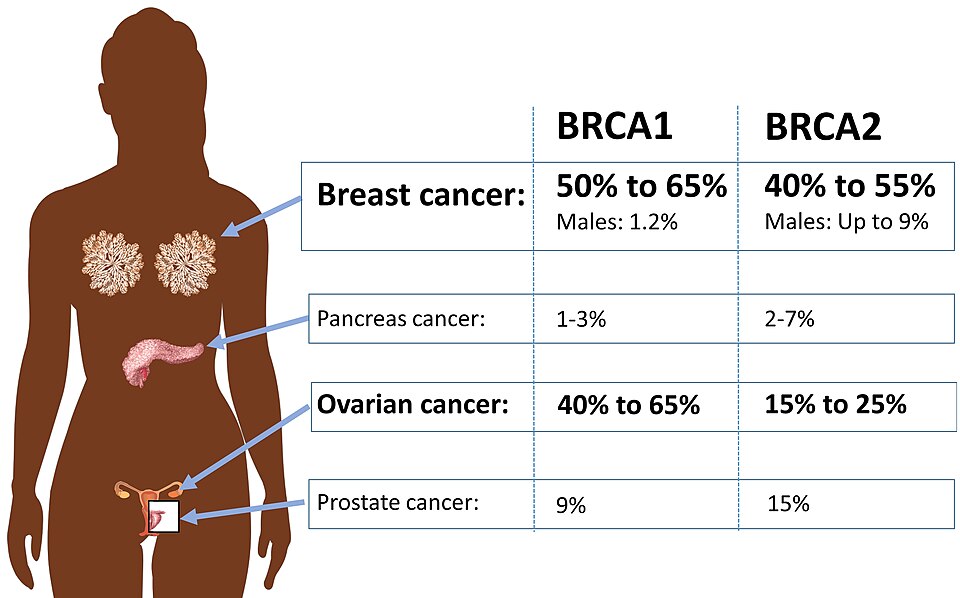
- BRCA1/2 mutations: autosomal dominant, loss of heterozygosity drives tumor. BRCA1: breast, ovarian, pancreatic. BRCA2: breast (male and female), ovarian, pancreatic, prostate, melanoma. BRCA2 = FANCD1 (Fanconi Anemia complementation group D1).
- Fanconi Anemia (FA): mutations in FA complementation genes (FANC genes). Aplastic anemia, skeletal anomalies (thumb abnormalities), short stature, increased AML and solid tumors. Cells are hypersensitive to DNA crosslinking agents (mitomycin C, diepoxybutane test).
Mnemonic: HR = "Homie Repairs" using a twin copy. S/G2 only. BRCA2 loads the RAD51 recombinase = BRCA2 is the loader.
03 · Disease Quick Map
Disease Quick Map
One table. Every repair disease. Scroll horizontally on mobile.
Disease
Defective Pathway
Enzyme / Gene
Key Finding
Xeroderma Pigmentosum
NER (global)
XPA through XPG, XPV
UV skin cancers in childhood, extreme sun sensitivity
Cockayne Syndrome
NER (TC-NER only)
CSA, CSB
Progeria-like aging, photosensitivity, no cancer
Lynch Syndrome (HNPCC)
Mismatch Repair (MMR)
MLH1, MSH2, MSH6, PMS2
Colorectal + endometrial CA, microsatellite instability (MSI-H)
BRCA1/2 Syndromes
Homologous Recombination (HR)
BRCA1, BRCA2
Breast + ovarian CA, autosomal dominant, BRCA2 also prostate/pancreatic
Fanconi Anemia
HR + crosslink repair
FANC genes (BRCA2 = FANCD1)
Aplastic anemia, thumb anomalies, AML, hypersensitivity to crosslinking agents
Absent T and B cells, radiosensitivity, V(D)J recombination failure
The unifying pattern: defective repair pathway accumulates mutations in a tissue-specific way. Which repair fails tells you which cancer. NER defects cause UV-driven skin cancers. MMR defects cause colorectal and endometrial cancers. HR defects cause breast and ovarian cancers. NHEJ defects cause immune deficiency (V(D)J fails).
XP vs. Cockayne distinction: Both are NER diseases, but XP loses global NER and gets skin cancers. Cockayne loses only TC-NER (the pathway that repairs damage in actively transcribed genes). Since bulk DNA repair still works, Cockayne patients do NOT get cancer. Instead they show photosensitivity, progeria-like aging, and neurological decline from failure to repair transcription-blocking lesions in neurons.
04 · Elimination Game
Elimination Game
Reveal clues one at a time. Eliminate wrong answers. Find the diagnosis.
A 25-year-old woman has a strong family history of colorectal cancer. Her father and paternal uncle were both diagnosed with colon cancer before age 50. Her aunt had endometrial cancer at 45. Genetic testing reveals she carries a pathogenic mutation in MSH2. She is currently healthy.
Which DNA repair process is deficient in this patient?
Nucleotide Excision Repair
Mismatch Repair
Homologous Recombination
Base Excision Repair
Clue 1: MSH2 is a key component of the MutS homolog complex. It recognizes mismatch bulges (wrong bases paired after DNA replication) and insertion/deletion loops at repetitive sequences. This function is shared with MSH6 in the MutSalpha complex.
This eliminates Nucleotide Excision Repair (NER repairs bulky UV lesions, uses XP gene products) and Base Excision Repair (BER uses glycosylases to remove small modified bases, e.g., MUTYH).
Clue 2: The family history in this vignette, colorectal cancer before 50 in multiple relatives plus endometrial cancer in a family member, is the textbook presentation of Lynch Syndrome (HNPCC). Lynch Syndrome is caused by germline mutations in MLH1, MSH2, MSH6, or PMS2, all proteins in the Mismatch Repair pathway.
This eliminates Homologous Recombination (HR defects cause breast and ovarian cancer via BRCA1/2, not colon and endometrial cancer).
Mismatch Repair (MMR) is deficient. MSH2 is an MMR protein. Lynch Syndrome (HNPCC) is the diagnosis. Screen for colorectal AND endometrial cancer.
05 · Retrieval Practice
Quiz
Four clinical questions. Original vignettes. Cover the choices first.
Question 1 of 4
A 10-year-old girl has a history of multiple squamous cell carcinomas of the skin. Her parents report she has always blistered with any sun exposure, even through window glass. On exam there are numerous pigmented macules with irregular borders scattered across sun-exposed areas. Neurological exam is normal.
Which type of DNA lesion accumulates in this patient because it is NOT being repaired?
ABase deamination (cytosine converted to uracil)
BInsertion/deletion loops at microsatellite repeats
CPyrimidine dimers from ultraviolet radiation
DDouble-strand breaks at fragile sites
Correct: C
Xeroderma Pigmentosum. UV radiation (UVB specifically) causes adjacent pyrimidines (usually thymine-thymine) to form covalent cyclobutane rings called pyrimidine dimers. These are bulky, helix-distorting lesions that can only be repaired by Nucleotide Excision Repair (NER). XP patients have NER gene mutations (XPA-XPG), so UV dimers accumulate with every sun exposure and cause mutations driving skin cancers.
A (Base deamination): Good instinct on base-level DNA damage, because deamination does alter bases. But what scale of damage is it? A single small base change, the substrate for base excision repair, where a glycosylase flips out one base. Is a UV dimer a single-base tweak or a bulky helix kink requiring a 25-nucleotide patch? The latter, which is NER. Match the lesion to the crew. Single-base deamination is repaired by base excision repair; a bulky UV pyrimidine dimer is repaired by NER.
B (IDLs at microsatellites): Tempting because microsatellite instability sounds like accumulated error. But where do insertion-deletion loops come from? Polymerase slippage at repeats, corrected by mismatch repair, whose failure is Lynch syndrome in colon and endometrium. Does that match a sun-sensitive child with skin cancer? No. UV photoproducts are an NER problem. Insertion-deletion loops at microsatellites are a mismatch repair (Lynch) problem; UV pyrimidine dimers are an NER (xeroderma pigmentosum) problem.
D (Double-strand breaks): Good instinct on serious physical damage, but which crews fix a double-strand break? Homologous recombination and non-homologous end joining. Does UV light sever both strands across from each other, or weld two pyrimidines on one strand? It welds one strand into a bulky lesion, the NER substrate. Different damage, different crew. Double-strand breaks are repaired by HR or NHEJ; bulky single-strand UV dimers are repaired by NER.
Question 2 of 4
A 44-year-old woman is newly diagnosed with colorectal cancer. Her oncologist orders tumor testing, which shows microsatellite instability-high (MSI-H) and loss of MLH1 protein expression by immunohistochemistry. She reports her mother had colorectal cancer at age 48 and her maternal grandmother had endometrial cancer at age 52. Germline testing confirms an MLH1 pathogenic variant.
Beyond colonoscopy surveillance, which additional cancer screening is highest priority for this patient?
AAnnual endometrial sampling or gynecologic surveillance
BAnnual mammography starting at age 30
CAnnual skin exam with dermatologist for melanoma
DAnnual low-dose CT scan of the chest for lung cancer
Correct: A
Lynch Syndrome (HNPCC) caused by MLH1 mutation. After colorectal cancer, endometrial cancer is the second most common malignancy in Lynch Syndrome, with lifetime risk of 40-60% in women. Mismatch repair deficiency allows replication errors to accumulate in rapidly dividing endometrial cells. Annual endometrial sampling and transvaginal ultrasound (or prophylactic hysterectomy) are guideline-recommended.
B (Mammography from age 30): Tempting because breast cancer is the first hereditary cancer most people associate with genetic testing. But which syndrome puts mammography from age 30 on the calendar? BRCA1/2, an HR defect. Does this MLH1 patient have an HR defect, or a mismatch repair defect? Mismatch repair, which targets colon, endometrium, and ovary, not breast. Screen the organ the mutation actually threatens. BRCA1/2 surveillance starts breast imaging early; Lynch (mismatch repair) surveillance prioritizes the endometrium, so this is the wrong syndrome's protocol.
C (Skin melanoma surveillance): Good instinct, since a few inherited syndromes do raise melanoma risk. But which ones? BRCA2 and CDKN2A carriers, not MLH1. Where does a mismatch repair defect strike? High-turnover epithelium: gut and endometrium, not stable melanocytes. The mutation here does not meaningfully raise melanoma risk. Melanoma surveillance fits BRCA2 or CDKN2A; an MLH1 Lynch patient is screened for endometrial, not melanoma, risk.
D (Lung CT for lung cancer): Who gets low-dose lung CT? Heavy smokers with high absolute lung cancer risk, not Lynch patients. Does mismatch repair failure target the lung? No, it targets colon and uterus. Match surveillance to the organ the mutation threatens, and the lung is not on the Lynch list. Lung CT screens smokers; Lynch surveillance is colonoscopy plus endometrial sampling, because mismatch repair failure hits the gut and uterus.
Question 3 of 4
A 38-year-old woman is found to carry a germline pathogenic variant in BRCA2. She has no current malignancy. Her oncologist explains that she has an elevated lifetime risk of breast and ovarian cancer and that her tumor suppressor function in DNA repair is impaired.
BRCA2 is required for which DNA repair pathway?
ANucleotide excision repair
BMismatch repair
CNon-homologous end joining
DHomologous recombination
Correct: D
BRCA2 is a central component of Homologous Recombination (HR). After a double-strand break in S or G2 phase, the broken ends are resected to expose single-stranded DNA. BRCA2's specific function is to load RAD51 recombinase onto these single-stranded 3' tails. RAD51 then catalyzes strand invasion into the sister chromatid for error-free repair. Without BRCA2, RAD51 cannot be properly loaded and HR is severely impaired, forcing the cell to use error-prone NHEJ instead.
A (Nucleotide excision repair): Good instinct that BRCA2 is a DNA repair protein, so any repair pathway feels plausible. But what lesion does NER handle? Bulky helix-distorting lesions like UV dimers, not broken strands. Is BRCA2 called for a warped base or for a snapped chromosome? A snapped chromosome, the double-strand break. Different damage, different crew. NER removes bulky lesions like UV dimers (the XP pathway); BRCA2 loads RAD51 for high-fidelity HR repair of double-strand breaks.
B (Mismatch repair): Tempting because mismatch repair is another cancer-predisposition pathway. But what does it correct? Base mismatches and slippage loops after replication, and BRCA2 plays no part in it. When is BRCA2 summoned? Only when both strands break. Match BRCA2 to the double-strand break, not the post-replication typo. Mismatch repair fixes replication mismatches (Lynch, microsatellite instability); BRCA2 enables HR repair of double-strand breaks and predisposes to breast and ovarian cancer.
C (Non-homologous end joining): Good instinct, since NHEJ is one of the two double-strand break pathways, so you are in the right damage category. But does NHEJ need BRCA2? No, it ligates ends without a template. BRCA2 is specific to the other branch, homologous recombination, which copies the intact sister chromatid for a faithful repair. Right damage, wrong branch. NHEJ is template-free and BRCA2-independent; HR is the high-fidelity branch that needs BRCA2 to load RAD51 onto resected ends.
Clinical corollary: BRCA2 = FANCD1 (Fanconi Anemia complementation group D1). Homozygous BRCA2 mutations cause Fanconi Anemia phenotype (aplastic anemia, AML) in childhood. Heterozygous mutations cause breast/ovarian cancer predisposition in adulthood.
Question 4 of 4
A researcher is studying telomerase activity across different human cell types. She finds that telomerase is highly active in a panel of tumor cell lines and in embryonic stem cells, but is undetectable in samples from differentiated skin fibroblasts taken from a healthy 35-year-old volunteer.
What is the best explanation for why telomerase is active in cancer cells but silent in normal somatic cells?
ACancer cells have a frameshift mutation in the TERT gene that activates enzyme activity
BNormal somatic cells lack the RNA template component (TERC) of telomerase
CCancer cells reactivate telomerase (most commonly via TERT promoter mutation or amplification) to prevent telomere shortening and achieve replicative immortality
DSomatic cells downregulate all reverse transcriptase activity to prevent retroviral replication
Correct: C
Telomerase is a reverse transcriptase that uses its own RNA template (TERC) to add TTAGGG repeats to chromosome ends. In normal somatic cells, the TERT gene (catalytic subunit) is epigenetically silenced after development. Without telomerase, chromosomes shorten with each replication cycle, triggering senescence or apoptosis. This is a built-in tumor suppressor mechanism.
Cancer cells overcome this by reactivating TERT, most commonly through TERT promoter mutations (creating new transcription factor binding sites), TERT amplification, or ALT (Alternative Lengthening of Telomeres).
A (Frameshift in TERT gene): Good instinct, since TERT reactivation is the mechanism, so a mutation in TERT feels right. But where does that reactivation happen, the coding sequence or the promoter? Mostly the promoter, where new transcription-factor binding sites switch the gene back on. A coding frameshift would break the enzyme, not re-express a working one. The cell needs a functional enzyme turned back on, which is a regulatory change. Cancer reactivates TERT mainly through promoter mutations or amplification, re-expressing a functional enzyme, not through coding frameshifts.
B (Normal cells lack TERC): Tempting because the RNA template TERC sounds like the missing piece. But is TERC actually scarce in somatic cells? No, it is broadly expressed. Which component is the silenced, rate-limiting one? TERT, the catalytic subunit. The blueprint is on the shelf; the licensed builder is what somatic cells withhold. TERC is widely expressed; somatic cells silence the catalytic subunit TERT, and cancers reactivate TERT, not re-acquire TERC.
D (Antiviral reverse transcriptase silencing): Interesting framing, but ask what silencing TERT actually accomplishes. It stops telomere maintenance so cells eventually senesce, a tumor-suppressor brake. Does that have anything to do with blocking retroviruses? No, those are separate regulatory circuits. The purpose of TERT silencing is preventing immortality, not viral defense. TERT silencing is a tumor-suppressor brake against replicative immortality; antiviral defense is an unrelated system.
0/4
quiz complete
06 · Clinical Gallery and Pearls
See It, Then Lock It
The faces of failed repair, the attending one-liner, and the hooks that make it stick. Tap any image to expand.
Xeroderma Pigmentosum NER lost: UV dimers persist. Tap to expand.
Ataxia-Telangiectasia ATM lost: dont X-ray them. Tap to expand.
UV Thymine Dimer The bulky lesion NER removes. Tap to expand.
Replication Fork Leading vs lagging, Okazaki. Tap to expand.
BRCA1/2 Risk HR loss: breast and ovarian. Tap to expand.
From the Attending
Stop memorizing diseases. Memorize the lesion. UV makes a bulky dimer, only NER pulls it out, lose NER and you get xeroderma pigmentosum. Every time. Ionizing radiation snaps both strands, ATM is the alarm that senses the snap, kill the alarm and you get ataxia-telangiectasia, so never order a routine film on that kid. Replication slippage at repeats is a mismatch problem, lose the MutS or MutL crew and the colon and endometrium light up: that is Lynch. Match the lesion to the crew and the diagnosis falls out on its own.
Memory Hook · NERNobody Escapes Radiation: NER clears UV bulky dimers. Lose global-genome NER and skin cancer comes for the XP child; lose only transcription-coupled NER and you get Cockayne (aging, no cancer).Tap to reveal
Memory Hook · MMR and LynchMismatch = Must catch Misspellings: the four Lynch genes are MLH1, MSH2, MSH6, PMS2. Loss gives microsatellite instability (MSI-H) and colon plus endometrial cancer.Tap to reveal
Memory Hook · HR and BRCABRCA2 is the loader: it loads RAD51 onto resected ends for homologous recombination. No HR backup plus a PARP inhibitor equals synthetic lethality, the engine of BRCA-targeted therapy.Tap to reveal
Memory Hook · Double-Strand BreaksHR is for S/G2, NHEJ is for G1: HR copies the sister chromatid (error-free); NHEJ tapes ends without a template (error-prone, and the basis of V(D)J). ATM is the kinase that sounds the alarm.Tap to reveal
Walk the chain on the XP child. Tap each beat to reveal the next link, then read the rule it builds to.
What does UV light physically do to DNA?
It fuses two adjacent pyrimidines on the same strand into a covalent cyclobutane ring, a bulky pyrimidine dimer that kinks the helix.
Which repair crew is built to excise a bulky helix distortion?
Nucleotide excision repair (NER): it cuts the strand on both sides of the lesion and replaces a 25 to 30 nucleotide patch.
A child loses the genome-wide arm of that crew. What is the syndrome?
Xeroderma pigmentosum, from loss of global-genome NER (XPA through XPG), with UV skin cancers in childhood.
What happens if only the transcription-coupled arm (CSA/CSB) is lost instead?
Cockayne syndrome: premature aging and neurodegeneration but no cancer, because bulk genomic repair still scrubs the mutagenic load.
The rule: UV makes bulky dimers, only NER removes them. Lose global-genome NER and you get xeroderma pigmentosum with childhood skin cancer; lose only transcription-coupled NER and you get Cockayne without cancer.
clinical Walkthrough
clinical Walkthrough
Original third-order vignettes. Shuffled, never-repeat, with full reasoning chains.
Medically reviewed by Kaitlyn Cocuzzo, MD and Fatima Ali, DO · Last updated July 1, 2026 at 10:03 PM ET
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.