Checkpoints, CDK/cyclin pairs, and the molecular brakes that stop cancer
A 55-year-old man with a 40-pack-year smoking history presents with hemoptysis. CT chest shows a 3.2 cm spiculated right upper lobe mass. Biopsy reveals adenocarcinoma. Molecular testing shows loss of p53 function and a constitutively active cyclin D1. Which cell cycle checkpoint is MOST directly impaired in this tumor?
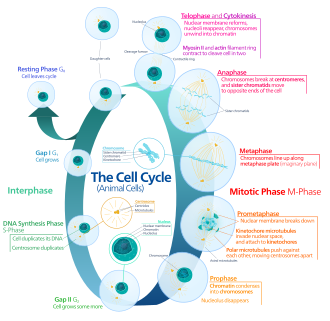
Four phases, one quiescent siding, four CDK/cyclin pairs. One wrong checkpoint and you get cancer.
Tap any image to enlarge. Cell-cycle map, mitotic stages, leukocoria of retinoblastoma, follicular lymphoma centrocytes, and a cell blebbing into apoptotic bodies.
The restriction point is the only door that matters. Past it, the cell does not look back: pull the mitogens and it still finishes the round. So when a stem hands you a stuck cyclin D1 or a dead p53 with no spindle clue, do not get cute about G2/M. Constitutive cyclin D1 or lost p53 with E2F running free is the G1/S restriction point, every time.
Gap 1 · Cell Growth
The cell grows, synthesizes proteins, and prepares for DNA replication. The most critical gate in the entire cycle is here: the G1/S checkpoint, sometimes called the "restriction point."
The Rb/E2F switch: Rb (retinoblastoma protein) acts as a brake. In its unphosphorylated form, Rb binds and sequesters the transcription factor E2F. When CDK4/6-Cyclin D phosphorylates Rb, it releases E2F, which then drives expression of genes needed for S phase entry (cyclins, DNA polymerases, etc.).
The brakes on the CDKs: p16 (encoded by CDKN2A) directly inhibits CDK4/6, preventing Rb phosphorylation. p21 (activated by p53 in response to DNA damage) inhibits both CDK2 and CDK4, keeping the cell arrested in G1 until damage is repaired.
DNA Synthesis
The entire genome is replicated exactly once. Two CDK/cyclin pairs cover the transition and progression.
CDK2 + Cyclin E: drives the G1 to S transition. Cyclin E levels peak right at the G1/S border, giving the final push through the restriction point.
CDK2 + Cyclin A: takes over during S phase progression. Cyclin A accumulates as S phase proceeds and keeps CDK2 active throughout replication.
Checkpoint during S: replication forks are monitored for stalling. ATR kinase senses single-stranded DNA at stalled forks and activates Chk1, which in turn inhibits CDK1/2, pausing progression until the block is resolved.
Gap 2 · Quality Check
The cell continues growing and verifies that DNA synthesis is complete and faithful. The G2/M checkpoint is the final quality inspection before committing to mitosis.
CDK1 + Cyclin B: this pair (also called MPF, Maturation Promoting Factor) is the engine of mitosis. It begins accumulating in G2 but remains inactive (held by Wee1 kinase phosphorylation on CDK1 Tyr15). The G2/M checkpoint is basically: is it safe to activate MPF?
Activation: Cdc25 phosphatase removes the inhibitory phosphate from CDK1, allowing CDK1/Cyclin B to fire and drive the cell into M phase. DNA damage activates ATM/ATR, which phosphorylate and inactivate Cdc25, blocking the G2/M transition.
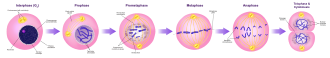
Mitosis · Division
PMAT: Prophase, Metaphase, Anaphase, Telophase, then cytokinesis. MPF (CDK1/Cyclin B) drives the cell through prophase into metaphase.
Spindle assembly checkpoint (SAC): the final checkpoint before anaphase. All chromosomes must be attached to spindle fibers from both poles (biorientation) before the cell is allowed to proceed. The SAC is mediated by the MCC (Mitotic Checkpoint Complex), which inhibits APC/C until every kinetochore is attached. A single unattached kinetochore is enough to halt the entire cell in metaphase.
Exiting mitosis: once all chromosomes are attached, APC/C (Anaphase Promoting Complex/Cyclosome) is activated. APC/C is an E3 ubiquitin ligase that ubiquitinates securin (releases separase, cleaves cohesins, allows sister chromatid separation) and Cyclin B (destroys it, inactivating CDK1, ending M phase).
Board pairs to lock in: cyclin D, CDK 4 the doorCyclin D pairs with CDK4/6 in G1 and phosphorylates Rb, opening the door to S phase.. CycE/CDK2 at the G1/S border, CycA/CDK2 through S phase, CycB/CDK1 (MPF) in G2 and M. Cyclin levels oscillate each cycle; CDK levels stay flat. A naked CDK does nothing without its cyclin partner.
Quiescence is a parking lot, not a death sentence. Whether a cell can drive back onto the cycle decides how a tissue heals, and p53 decides whether a damaged cell lives at all.
A cell that stops dividing exits G1 into G0G0 is a resting, quiescent state outside the cycle. Cells sit here doing their job; some can re-enter G1 when signaled, some never can., a quiescent siding off the main loop. Mitogens (growth factor to receptor tyrosine kinase to RAS to cyclin D) call a cell back into G1. Whether it can answer that call is fixed by tissue type:
Students love to say the heart heals. It does not. Tempting because the patient survives the MI, but survival is scar, not myocardium. Neurons, cardiac, and skeletal muscle are permanent: dead means replaced by fibrosis, never by new cells.
The single most mutated gene in human cancer, and the bridge from a broken checkpoint to the intrinsic apoptosis program.
Lose both TP53 alleles and that whole chain breaks: damaged cells neither arrest nor die, and mutations stack. Germline loss of one allele is Li-Fraumeni syndrome (sarcomas, breast cancer, brain tumors, adrenocortical carcinoma at young ages). p53 is the guardian of the genome: it arrests damaged cells via p21 and, if repair fails, executes them through the intrinsic pathway.
Gas pedals that get stuck vs. brakes that break. Both cause cancer, opposite mechanisms.
| Gene | Class | Associated Cancer(s) | Notes |
|---|---|---|---|
| KRAS | Oncogene | Pancreatic (90%), colon, lung adenocarcinoma | GTPase locked in "on" state; can't hydrolyze GTP to GDP |
| MYC | Oncogene | Burkitt lymphoma, small cell lung cancer | t(8;14) in Burkitt: MYC moves next to IgH promoter |
| BCR-ABL | Oncogene | CML, some ALL | t(9;22) Philadelphia chromosome; constitutively active tyrosine kinase; targeted by imatinib |
| HER2 | Oncogene | Breast (20-25%), gastric | Gene amplification (not point mutation); targeted by trastuzumab |
| TP53 | TSG | Li-Fraumeni, most human cancers | Li-Fraumeni: germline TP53 mutation; sarcomas, brain tumors, breast, leukemias |
| RB1 | TSG | Retinoblastoma, osteosarcoma, small cell lung | Knudson two-hit hypothesis first demonstrated here; chromosome 13q14 |
| BRCA1 | TSG | Breast, ovarian | DNA double-strand break repair; BRCA2 also involved; both autosomal dominant inheritance |
| APC | TSG | FAP, colorectal | FAP: germline APC mutation; >100 colonic polyps by 2nd decade; colon cancer by 40s |
Two roads to the same executioners. Know which caspases sit where and what triggers them.
Mitochondrial Pathway
Triggered by internal stress signals: DNA damage, hypoxia, reactive oxygen species, oncogene activation, growth factor withdrawal. The mitochondria are the decision hub.
BCL-2 family proteins control the gate:
· Anti-apoptotic (guards): BCL-2, BCL-XL, MCL-1. They keep the outer mitochondrial membrane intact.
· Pro-apoptotic (killers): BAX, BAK. They form pores in the outer membrane.
· BH3-only sensors: BAD, BIM, PUMA, NOXA. Activated by stress, they neutralize BCL-2/BCL-XL, freeing BAX/BAK to punch holes.
When BAX/BAK oligomerize, they cause mitochondrial outer membrane permeabilization (MOMP), releasing cytochrome c into the cytoplasm. Cytochrome c binds Apaf-1, which recruits procaspase-9 to form the apoptosome. Caspase-9 activates caspase-3 (executioner).
Death Receptor Pathway
Triggered from outside the cell by ligands binding death receptors on the plasma membrane. Two main axes:
Fas-FasL: FasL (on cytotoxic T cells or other cells) binds Fas receptor (CD95) on the target cell. Fas trimerizes and recruits FADD (Fas-Associated Death Domain) adapter protein via its death domain. FADD recruits procaspase-8, forming the DISC (Death-Inducing Signaling Complex). Procaspase-8 autoactivates to caspase-8, which directly cleaves and activates procaspase-3.
TNF-TNFR1: TNF binds TNFR1, recruits TRADD (TNFR-Associated Death Domain), then FADD, then caspase-8. Can also activate NF-kB (survival) depending on context.
Granzyme B: cytotoxic T cells and NK cells release perforin (forms pores in target membrane) and granzyme B (serine protease). Granzyme B enters the target cell and directly cleaves/activates caspase-3, bypassing the receptor-FADD-caspase-8 cascade entirely.
Final Common Path
Both intrinsic (caspase-9) and extrinsic (caspase-8) pathways converge on the executioner caspases: caspase-3, caspase-6, and caspase-7. Once activated, these proteases systematically dismantle the cell.
What they cleave:
· PARP (poly-ADP ribose polymerase): disables DNA repair, commits cell to death
· Lamin A/B (nuclear lamins): nuclear envelope collapses
· Cytoskeletal proteins (fodrin, actin): cell shrinks
· CAD inhibitor (ICAD): activates the DNase CAD, which cleaves DNA into oligonucleosomal fragments (the "DNA ladder" on gel electrophoresis)
Morphology of apoptosis: cell shrinks, chromatin condenses, membrane blebs, nucleus fragments, cell breaks into apoptotic bodies (membrane-enclosed packages). Phagocytes recognize phosphatidylserine flipped to the outer leaflet and eat the packages. No inflammation because contents are never released.
Four candidates. Two clues. Eliminate wrong answers as evidence arrives.
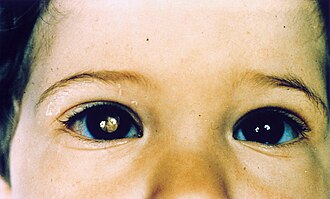
The gene is the founding example of the two-hit hypothesis for tumor suppressor genes. Alfred Knudson described this exact cancer when proposing that both copies of a gene must be lost before a tumor forms. Hereditary cases present earlier and bilaterally; sporadic cases are unilateral.
The protein product normally binds and sequesters E2F transcription factor when unphosphorylated. When CDK4/6-Cyclin D phosphorylates it, E2F is released and drives S phase entry. In tumors lacking this protein, E2F is constitutively free and the cell cannot be held in G1.
Original third-order vignettes, one at a time. Lock your answer, then tap each beat to walk the reasoning. Long-press or right-click an option to cross it out; double-tap to highlight.