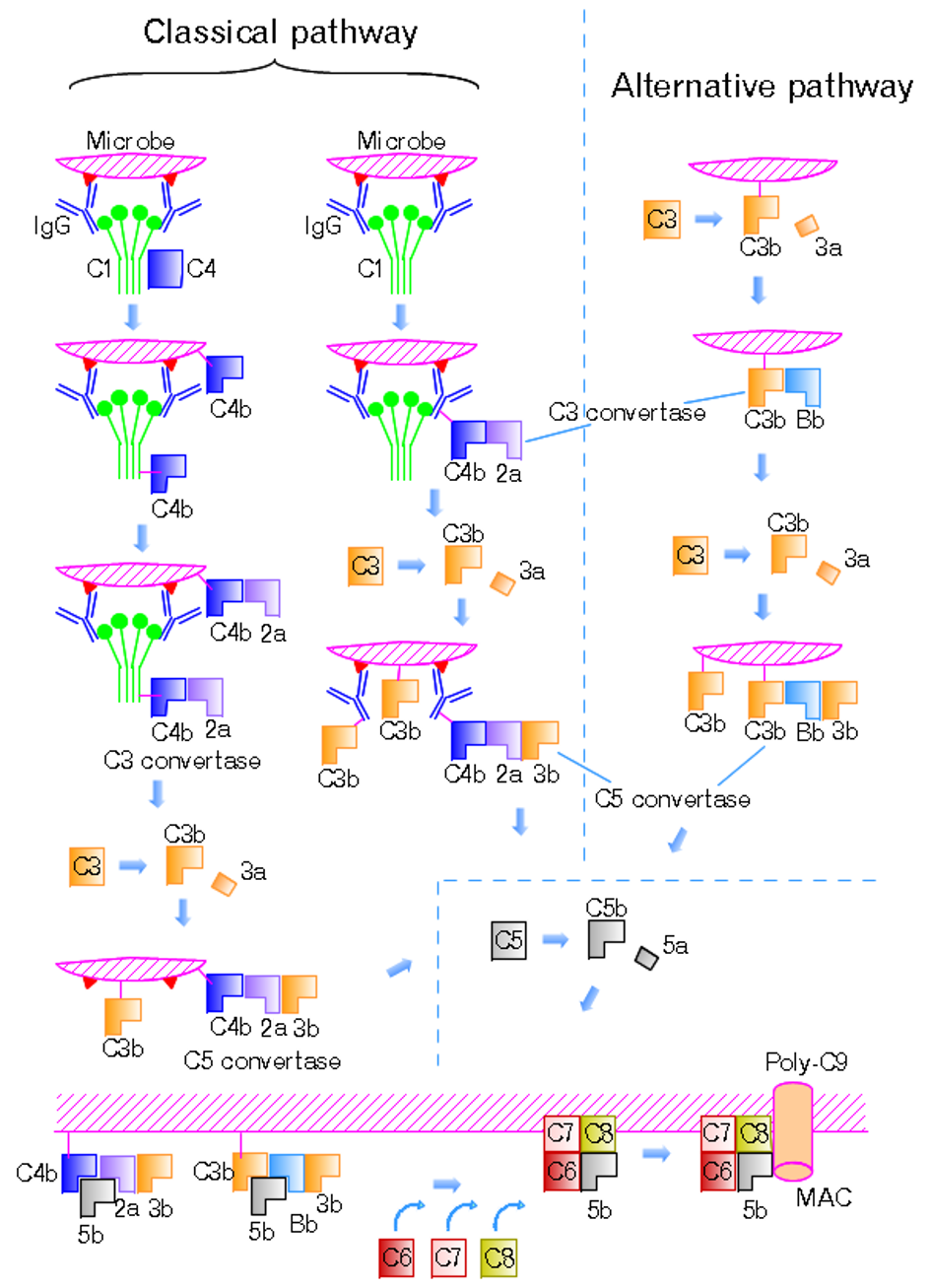
You already know the cascade fires. This page is the other half: what happens to a patient when one piece is missing or one brake is gone. Each defect has a syndrome, a lab signature, and a treatment the board loves to test.
A 34-year-old woman has her third episode this year of painless swelling of the lips and tongue lasting two days. There is no hives and no itch, and she had two relatives with the same problem. Antihistamines and epinephrine never helped. Which feature most reliably tells you this is hereditary angioedema and not an allergic reaction?
It swells the face
No urticaria and no itch
It lasts only minutes
It responds to epinephrine
Allergic angioedema is a histamine story: mast cells dump histamine, so you get hives, itch, and flushing alongside the swelling, and it answers to epinephrine and antihistamines. Hereditary angioedema is a bradykinin story: there is no mast cell, so no hives and no itch, just deep swelling, and the usual allergy drugs do nothing. That single line, swelling without urticaria, is the discriminator. Underneath it sits a low C4 and a broken C1 inhibitor. No hives plus no response to epinephrine equals bradykinin, equals hereditary angioedema.
Begin
One Cascade, Many Failure Points
Where Is the Defect?
This page assumes you know how the cascade fires. Here we map the breakages. Tap any glowing component or regulator and the panel tells you what its loss does to the patient.
Three pathways (classical, lectin, alternative) converge on C3, then drive C5 to C9 into the membrane attack complex. Every syndrome on this page is a break somewhere along this road.
Classical C1 · C2 · C4
Lectin MBL · MASP
Alternative C3b · B · D
C1 inhibitor the brake on C1
Factor H / I brake on C3b
Properdin props up C3bBb
▼
C3 the hub
DAF (CD55) + CD59 surface brakes
▼
MAC: C5 to C9 the hole punch
Tap a component
Yellow tags mark a clickable break. Tap one and this panel shows what it normally does, the syndrome its loss causes, and the lab pattern that gives it away.
The whole page in one rule
Lose a component and you get too little defense (infections, immune complexes). Lose a brake and you get too much activation (swelling, hemolysis, microangiopathy). Sort every deficiency into one of those two buckets first.
🧠Missing soldier equals infection. Missing brake equals self-damage. Two buckets, every time.
Lost Regulators, Runaway Activity
Brakes Off: HAE & PNH
Two diseases of missing brakes. C1 inhibitor restrains bradykinin; lose it and you swell. DAF and CD59 protect your own red cells from the membrane attack complex; lose them and the cells burst from inside the vessel.
Angioedema · deep swelling, no hives
HAE attack · non-pitting hand swelling
PNH · dark hemoglobin in the urine
Brake lost
😭
Hereditary Angioedema
C1 inhibitor deficiency (autosomal dominant)
MediatorBradykinin (not histamine)
Screen labLow C4 always
AvoidACE inhibitors
🔍 Swelling of face, larynx, gut with no hives and no itch
tap to flip →
HAE up close
PresentationRecurrent deep swelling of the face, lips, larynx, and gut wall. GI attacks mimic an acute abdomen with colicky pain. Laryngeal edema can be fatal. No urticaria, no pruritus. Type 1 (85 percent): low C1-INH level. Type 2: normal level, broken protein.
Labs / levelsLow C4 at all times, even between attacks, so it screens. Low C1-INH level or function. C1q is normal in hereditary disease; low C1q points to acquired C1-INH deficiency with a lymphoproliferative or autoimmune cause.
TreatmentAcute: C1-INH concentrate, icatibant (bradykinin B2 blocker), ecallantide (kallikrein blocker). Prophylaxis: berotralstat, danazol, lanadelumab. Antihistamines, epinephrine, and steroids do not reliably work.
Brake lost
🩸
PNH
DAF (CD55) + CD59 loss, acquired PIGA mutation
HemolysisIntravascular
CoombsNegative
DiagnoseFlow cytometry
🔍 Dark morning urine, thrombosis in odd veins, marrow failure
tap to flip →
PNH up close
PresentationAn acquired PIGAA gene on the X chromosome needed to build the GPI anchor. A mutation in one stem cell makes a clone of blood cells with no GPI anchor, so they lose the surface brakes DAF and CD59. mutation removes the GPI anchor, so cells lose DAF and CD59 and complement lyses them inside the vessel. Episodic dark urine, classically morning or night. Venous thrombosis in odd sites (hepatic vein, Budd-Chiari) is the leading cause of death. Overlaps with marrow failure and pancytopenia.
Labs / levelsHigh LDH, low haptoglobin, hemoglobinuria, hemosiderinuria. Direct Coombs is negative because complement, not antibody, kills the cell.
Diagnosis / treatmentFlow cytometry shows absent CD55 and CD59 (FLAER assay). Treat with eculizumab or ravulizumab (anti-C5). Vaccinate against Neisseria first; anticoagulate for clots. Stem cell transplant is curative.
Why HAE has a low C4 even when she feels fine
C1 inhibitor is the brake on activated C1. With the brake gone, C1 keeps chewing through C4 and C2 around the clock, so C4 stays low all the time. That makes a resting low C4 the cheap, always-abnormal screen.
🧠No brake on C1 means C1 eats C4 nonstop. Low C4 even between attacks.
The bradykinin trap
Because HAE is bradykinin, not histamine, antihistamines, epinephrine, and corticosteroids do not reliably work, and ACE inhibitors are contraindicated (ACE normally breaks bradykinin down). If a stem treats angioedema like anaphylaxis and it fails, think C1 inhibitor.
Challenge: Sort the Angioedema
Commit before you peek. Two linked nodes drill the allergic versus hereditary split and then the treatment.
Step 1. A patient has recurrent deep facial and tongue swelling with no hives, no itch, and a family history. Allergic reaction or hereditary angioedema?
Allergic (histamine) angioedema
Hereditary angioedema
Step 2. She is now having a laryngeal attack in front of you. Which is the right acute move?
Load up on antihistamines and steroids
C1 inhibitor concentrate or icatibant
Missing Components, Open Doors
Too Little: Neisseria, Pyogenic, Lupus-like
Lose an actual component and the body cannot finish a job. The terminal complex cannot lyse Neisseria, C3 cannot opsonize, and the early classical pieces cannot clear immune complexes. Three different doors swing open.
Petechiae and purpura of meningococcemia. A second episode of Neisseria infection is the classic flag for a terminal or properdin defect.
Component lost
🦠
Terminal & Properdin
C5 to C9 (MAC) or properdin deficiency
RiskRecurrent Neisseria
ScreenLow CH50
BugsMeningococcus, gonococcus
🔍 A second meningococcal infection equals a terminal complement defect
tap to flip →
Terminal / properdin up close
PresentationRecurrent or unusually severe Neisseria infection (meningococcal meningitis, disseminated gonococcus). The MAC punches holes in the thin Neisseria wall, so without C5 to C9 it cannot be killed. Properdin (X-linked) stabilizes the alternative pathway convertase; its loss gives fulminant meningococcemia.
Labs / levelsLow or absent CH50A functional test of the whole classical plus terminal pathway, C1 through C9. A flat CH50 with normal early labs points at a terminal component.. Individual terminal component is undetectable.
TreatmentMeningococcal vaccination and prompt antibiotics for infections. Same logic explains why eculizumab, which blocks C5, demands meningococcal vaccination first.
Component lost
🧬
C3 Deficiency
Loss of the central hub C3
RiskSevere pyogenic infection
BugsEncapsulated organisms
AlsoImmune complex disease
🔍 The hub is gone, so opsonization and the whole downstream fail
tap to flip →
C3 deficiency up close
PresentationSevere, recurrent pyogenic infections with encapsulated organisms (pneumococcus, H. influenzae, meningococcus), because C3b is the main opsonin. Also type III immune complex disease (glomerulonephritis) from poor clearance.
Labs / levelsLow C3. Because C3 sits at the convergence of all pathways, both CH50 and the alternative test are abnormal.
TreatmentAggressive vaccination against encapsulated organisms and prompt treatment of infections.
Component lost
🥖
Early Classical
C1, C2, or C4 deficiency
ClassicLupus-like disease
Most commonC2 deficiency
AlsoSinopulmonary infection
🔍 Cannot clear immune complexes, so it looks like lupus
tap to flip →
Early classical up close
PresentationA lupus-like picture: malar rash, photosensitivity, arthralgias. The early classical pieces clear immune complexes and dying cells, so losing them lets complexes deposit and inflame. Also recurrent sinopulmonary (encapsulated) infections.
Labs / levelsLow C4 and/or C2. C2 deficiency is the most common inherited complement deficiency.
TreatmentManage the autoimmune disease, vaccinate, and treat infections promptly.
Three doors in one lineTerminal / properdin opens the door to Neisseria. C3 opens the door to encapsulated pyogenic bugs. Early classical (C1, C2, C4) opens the door to lupus-like immune complex disease.
🧠Terminal goes to Neisseria. C3 goes to pus. Early classical goes to lupus.
Regulator Failure in the Glomerulus
The Kidney: aHUS & Dense Deposit Disease
When the alternative pathway brakes fail, the kidney pays. Factor H or Factor I loss drives a clot-and-shred microangiopathy. A rogue autoantibody that props up the C3 convertase burns C3 to the floor and scars the glomerulus.
Schistocytes · the shredded red cells of a microangiopathy
🔍 The same triad as typical HUS, but no diarrhea and driven by complement
tap to flip →
Factor H / I and aHUS up close
PresentationComplement-driven thrombotic microangiopathyTiny clots in small vessels that shred passing red cells and consume platelets, while starving the kidney of flow.: fragmented red cells (microangiopathic hemolytic anemia), thrombocytopenia, and acute kidney injury. No diarrheal prodrome; often familial and relapsing.
Labs / levelsSchistocytes on smear, low platelets, rising creatinine. C3 may be low; C4 is usually normal because the trouble is the alternative pathway. Genetics show Factor H or Factor I loss-of-function.
TreatmentEculizumab (anti-C5). Contrast with typical HUS, which follows E. coli O157:H7 and is managed supportively.
Brake propped open
🍳
Dense Deposit Disease
C3 nephritic factor, C3 glomerulopathy
C3Very low
C4Normal
PatternMPGN type II
🔍 An autoantibody jams the C3 convertase on, draining C3 to nothing
tap to flip →
Dense deposit disease up close
PresentationA nephritic and nephrotic mix (hematuria, proteinuria, edema, hypertension). The lesion is a membranoproliferative pattern (MPGN type II), now grouped under C3 glomerulopathy. Sometimes paired with partial lipodystrophy.
Labs / levelsC3 nephritic factorAn autoantibody that binds and stabilizes the alternative pathway C3 convertase (C3bBb), keeping it switched on so it shreds C3 continuously. stabilizes C3bBb, so the alternative pathway runs nonstop and C3 falls very low while C4 stays normal.
TreatmentSupportive nephrology care; complement-directed therapy is used in selected C3 glomerulopathy cases.
Same Triad, Two Stories
HUS is microangiopathic hemolytic anemia plus thrombocytopenia plus acute kidney injury. The split is what caused it. Tap each tab.
Typical HUS (Shiga)
Atypical HUS (complement)
Typical HUS · Shiga toxin
A child with a bloody diarrheal prodrome, classically from E. coli O157:H7 in undercooked beef (or Shigella).
Shiga toxin damages endothelium directly.
Management is supportive; antibiotics are generally avoided.
Normal complement levels.
Atypical HUS · complement
No diarrhea. Uncontrolled alternative pathway from Factor H or Factor I dysfunction, often familial and relapsing.
Complement attacks endothelium directly.
Treat with eculizumab.
C3 may be low; C4 usually normal.
Challenge: a 4-year-old has bloody diarrhea, then fragmented red cells, low platelets, and acute kidney injury. Most likely driver?
Shiga toxin (typical HUS)
Complement dysregulation (atypical HUS)
The low C3 fork
A persistently low C3 with a normal C4 means the trouble is the alternative pathway: think C3 nephritic factor and dense deposit disease, or Factor H / I disease. A low C4 instead points back at the classical pathway (HAE, early classical deficiency).
🧠Low C3, normal C4 equals alternative pathway. Low C4 equals classical pathway.
Read the Labs, Name the Defect
The Complement Decoder
A case card lands with a presentation and a lab line. Pick the deficiency that fits. Get it and the next card loads. This is exactly how the board hides the diagnosis inside the numbers.
Case 1 of 5
Detective: Which Deficiency?
Four deficiencies are on the table. Each clue rules one out. Tap the one the clue eliminates. Last card standing is the answer.
A patient has recurrent infection and you must place the complement defect. Read each clue, then tap the deficiency it eliminates.
Terminal C5 to C9
Neisseria
C1 inhibitor
angioedema
C3
pyogenic, encapsulated
Early classical
lupus-like
Loading clue...
Prove It
Clinical Walkthrough
Original full clinical vignettes, one at a time. Shuffled, never-repeat. Answer first, then every choice gets explained and the reasoning chain unlocks. Six patients walked in. Find the defect.
Lock It In
Rapid Fire Quiz
Five questions per load, shuffled from a larger bank. Wrong answers explain why the trap felt right, then hand you the rule. Reload for a fresh set.
References: standard immunology and clinical pathology board texts. Images: Wikimedia Commons (see each lightbox for attribution).
Medically reviewed by Fatima Ali, DO and Kaitlyn Cocuzzo, MD. Vignettes are original clinical teaching cases; demographics, values, and answer order are written for practice. Confirm management against current references at the point of care.
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.