The Four Leukemias
Acute vs. chronic. Myeloid vs. lymphoid. The 2x2 grid that organizes everything.


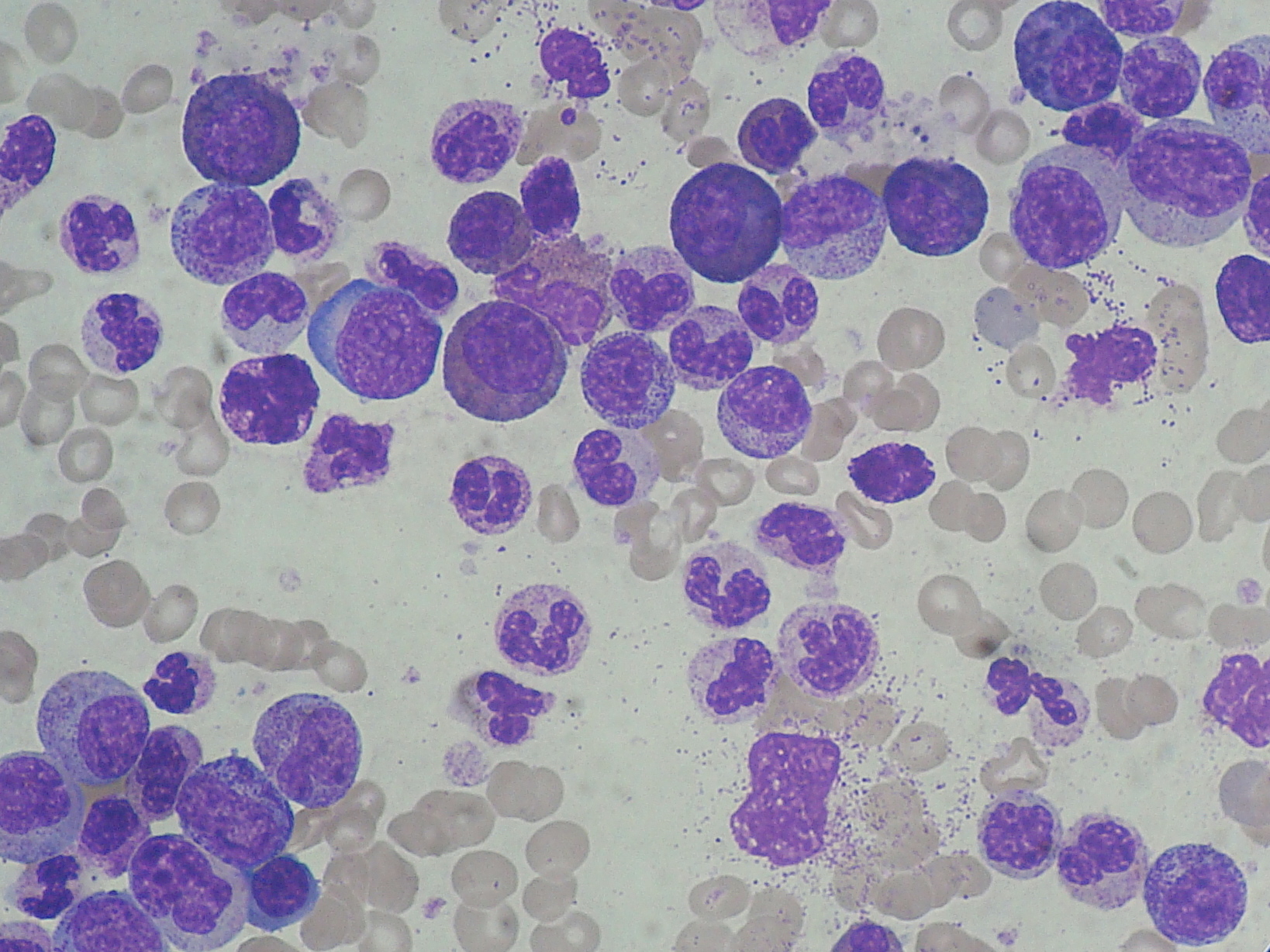

Acute Myeloid Leukemia
Adults · Median age 65 · Blasts from myeloid lineage
Diagnosis: Blasts >20% in bone marrow. Myeloperoxidase (MPO) positive. Surface markers: CD13, CD33, CD117. Auer rods in cytoplasm of blasts = pathognomonic for AML.
Most board-tested subtype: M3 (APL, Acute Promyelocytic Leukemia). Translocation t(15;17) fuses PML and RARA genes. The PML-RARA fusion protein blocks myeloid differentiation at the promyelocyte stage. Promyelocytes pack with granules that dump coagulation factors on cell death, causing DIC. Auer rods are most prominent in M3.
Treatment of M3: ATRA (all-trans retinoic acid) + arsenic trioxide. ATRA binds the RARA component of the fusion protein and forces terminal differentiation of the stuck promyelocytes. This is "differentiation therapy" not cytotoxic chemotherapy. The blasts mature into normal neutrophils instead of dying and releasing granules, which is why it also resolves the DIC risk.
Other AML causes: Prior radiation, prior alkylating agent chemo, topoisomerase II inhibitor chemo (balanced translocations), Down syndrome (21-fold increased risk), progression from myelodysplastic syndrome (MDS).
Standard AML treatment (non-M3): "7+3" induction: cytarabine for 7 days continuous infusion + anthracycline (daunorubicin or idarubicin) for 3 days. Allogeneic stem cell transplant in first remission for fit patients with intermediate/adverse risk cytogenetics.
Acute Lymphoblastic Leukemia
Children · Peak age 2-5 years · Most common childhood cancer
Diagnosis: Lymphoblasts. TdT+ (terminal deoxynucleotidyl transferase) is the hallmark of ALL; it marks immature lymphocytes. B-ALL (more common): CD10+ (CALLA), CD19+, CD20+. T-ALL: CD3+, CD7+, CD1a+.
Clinical presentation: Bone pain and limping (marrow replacement compresses periosteum), lymphadenopathy, pallor, bruising, petechiae. T-ALL specifically: mediastinal mass (thymic origin), can cause superior vena cava syndrome. CNS involvement: headache, diplopia, cranial nerve palsies (ALL has the highest CNS tropism of all leukemias).
High-risk cytogenetics: Philadelphia chromosome t(9;22), BCR-ABL fusion. This is the same translocation as CML but appearing in ALL confers terrible prognosis. Treatment response: add a TKI (imatinib or dasatinib) to the chemotherapy backbone.
Treatment: Multi-agent chemotherapy protocols (hyper-CVAD or Berlin-Frankfurt-Munster [BFM]). Total duration 2-3 years. CNS prophylaxis is mandatory: intrathecal methotrexate (IT-MTX) prevents CNS relapse. Cranial radiation avoided now due to late cognitive effects. Prognosis: childhood B-ALL approaches 90% long-term cure rate.
Chronic Myeloid Leukemia
Adults 40-60 years · Philadelphia chromosome · Tyrosine kinase story
Defining lesion: Philadelphia chromosome, t(9;22). The ABL1 gene from chromosome 9 fuses with BCR on chromosome 22. BCR-ABL1 encodes a constitutively active tyrosine kinase that drives uncontrolled myeloid proliferation. This is the founding oncogene story of targeted therapy.
Three phases: (1) Chronic phase: years long, mostly mature myeloid cells at all stages, leukocytosis often >100,000, splenomegaly (often massive), basophilia. Symptoms: fatigue, early satiety, left upper quadrant fullness. (2) Accelerated phase: blasts 10-19% in blood or marrow, increasing basophilia, worsening cytopenia. (3) Blast crisis: blasts ≥20%, behaves like acute leukemia (two-thirds myeloid, one-third lymphoid). Median survival in blast crisis historically weeks to months.
Key lab finding: Low leukocyte alkaline phosphatase (LAP) score. LAP is normally high in reactive leukocytosis (leukemoid reaction). CML cells are neoplastic and don't upregulate LAP. If WBC is 80,000 and you need to distinguish CML from a severe infection: low LAP = CML; high LAP = leukemoid reaction.
Treatment: Imatinib (Gleevec) was the original BCR-ABL TKI and transformed CML from a disease with median survival of 3-4 years into one where patients can achieve near-normal life expectancy. Second-generation TKIs (dasatinib, nilotinib) for resistance or intolerance. Ponatinib for T315I "gatekeeper" mutation that confers resistance to all other TKIs.
Chronic Lymphocytic Leukemia
Elderly >65 years · Most common adult leukemia in Western world · B-cell
Diagnosis: Monoclonal B-lymphocytes with characteristic immunophenotype: CD5+ / CD19+ / CD23+. CD5 is normally a T-cell marker; its aberrant expression on B-cells defines CLL. Smudge cells on peripheral smear: lymphocytes so fragile they rupture during slide preparation, leaving a chromatin smear. Lymphocytosis >5,000/mcL sustained for at least 3 months.
Clinical: Most patients are asymptomatic at diagnosis. Diffuse lymphadenopathy, splenomegaly, and hepatomegaly develop over time. The immunoglobulins are dysfunctional (hypogammaglobulinemia) despite the high lymphocyte count, so patients get recurrent sinopulmonary infections with encapsulated organisms.
Complications: (1) Autoimmune hemolytic anemia (warm AIHA): DAT (direct antiglobulin test) positive. CLL B-cells make autoantibodies against RBCs. (2) Richter transformation: 5-10% of CLL patients undergo sudden transformation to diffuse large B-cell lymphoma. Presents as rapidly enlarging nodes, B symptoms (fever, night sweats, weight loss), markedly elevated LDH. Biopsy confirms.
Staging: Rai staging: Stage 0 = lymphocytosis only. Stage I adds lymphadenopathy. Stage II adds organomegaly. Stage III adds anemia. Stage IV adds thrombocytopenia. Higher stage = worse prognosis.
Cytogenetics: del(13q) = best prognosis (most common abnormality in CLL). del(17p) = worst prognosis (p53 deletion, poor response to most therapies). del(11q) and +12 = intermediate.
Treatment: Asymptomatic patients: watch and wait (treatment does not improve survival in early CLL). Symptomatic: ibrutinib (BTK inhibitor), venetoclax (BCL-2 inhibitor), obinutuzumab (anti-CD20). Chlorambucil-based regimens still used in very elderly. Allo-SCT rare, reserved for young high-risk patients.
Age and Association
Four leukemias in one view. Scroll horizontally on mobile.
| Leukemia | Peak Age | Key Finding | Translocation | Treatment |
|---|---|---|---|---|
| AML (M3/APL) | Adults (median 65) | Auer rods, DIC, MPO+ | t(15;17) PML-RARA | ATRA + arsenic trioxide |
| ALL | Children 2-5 yrs | TdT+, CD10+, mediastinal mass (T-ALL) | t(9;22) Ph+ = worst prognosis | Multi-agent chemo + TKI if Ph+; IT-MTX for CNS |
| CML | Adults 40-60 yrs | Low LAP, basophilia, massive splenomegaly | t(9;22) BCR-ABL1 (defines disease) | Imatinib (TKI) first-line |
| CLL | Elderly >65 yrs | Smudge cells, CD5+/CD19+/CD23+, hypogammaglobulinemia | del(13q) best, del(17p) worst | Watch + wait (asymp.), ibrutinib/venetoclax (sympt.) |
Lab Findings Drill
Tap each card to expand. Know these cold before test day.
Auer rods: Pink, needle-shaped cytoplasmic inclusions in myeloblasts. Pathognomonic for AML. Most prominent in M3 (APL). Their presence in a blast = AML until proven otherwise.
Smudge cells: CLL. The neoplastic lymphocytes are so fragile that the mechanical force of spreading the smear ruptures them. The remnant chromatin forms a smear on the slide. Classic for CLL but not specific to it.
Lymphoblasts (ALL): Large cells with scant cytoplasm, prominent nucleoli, fine chromatin. TdT+. Distinguish from myeloblasts: ALL blasts are MPO-negative and TdT-positive; AML blasts are MPO-positive.
CML smear: Full myeloid spectrum all at once: myeloblasts, promyelocytes, myelocytes, metamyelocytes, bands, and mature neutrophils. Plus prominent basophilia and eosinophilia. Looks like a bone marrow aspirate ended up in the blood.
Myeloperoxidase (MPO) stain: Positive in myeloid lineage (AML). Negative in lymphoid lineage (ALL). The single most useful stain to distinguish AML from ALL when flow cytometry isn't immediately available.
Sudan black B: Same staining pattern as MPO. Stains lipids in myeloid granules. Positive in AML, negative in ALL.
TdT (terminal deoxynucleotidyl transferase): Nuclear enzyme that adds non-templated nucleotides during lymphocyte VDJ recombination. Marks immature lymphocytes. Positive in ALL and also in some AML cases (overlap). Key ALL marker.
PAS (periodic acid-Schiff): Stains glycogen. Positive in ALL lymphoblasts as large blocks of glycogen in the cytoplasm. This gives ALL a characteristic "PAS-positive blocks" appearance, useful when flow cytometry is unavailable.
t(15;17): APL / AML-M3. PML-RARA fusion. Blocked differentiation at promyelocyte stage. Respond to ATRA. DIC risk at diagnosis.
t(9;22): Appears in CML (defines the disease, BCR-ABL1) AND in ALL (Ph+ ALL, worst adult ALL). Different clinical context, same chromosomal event.
t(8;21): AML-M2. Good prognosis. RUNX1-RUNX1T1 fusion. Auer rods common. Young adults. High complete remission rate with standard chemo.
inv(16): AML-M4eo (myelomonocytic with eosinophilia). CBFB-MYH11 fusion. Good prognosis. Abnormal eosinophils in marrow.
del(17p) in CLL: Deletes TP53. Worst prognosis in CLL. Resistant to most chemo. Ibrutinib or venetoclax bypass the p53 pathway and remain effective.
del(13q) in CLL: Most common cytogenetic abnormality in CLL. Best prognosis. Watch-and-wait patients often have this alone.
Elimination Game
Read the vignette. Use the clues to eliminate one by one until only the answer remains.
Board Walkthrough
25 original board-style vignettes. One at a time, shuffled, never repeats. Right-click or long-press to cross out. Double-tap to highlight.